2.5 Self assembly using Rh and Sn coordination chemistry
A combination of Sn(IV) and Rh(III) porphyrin coordination chemistry seemed attractive for the self assembly of large ordered structures in solution and the solid state. As described in the preceding sections, Rh(III) porphyrin has an affinity for pyridyl ligands but binding of carboxylates is unknown, whereas Sn(IV) porphyrin is oxophilic in character and shows selectivity for binding of carboxylates.286-289 The porphyrins also contrast in the number of axial ligands bound. Sn(IV) ligates up to two carboxylates, and can thus act as a ‘core’ for bringing together two carboxylic acid functionalized components. Rh(III)-halo porphyrin, as prepared, binds a single ligand readily, in exchange for a weakly coordinated solvent molecule. However the halide ligand of Rh(III) halo porphyrins is usually difficult to displace, unless more forcing conditions are used such as photolysis or treatment with Ag+.290 Both types of metalloporphyrins are readily investigated by NMR spectroscopy, on account of their diamagnetic nature, slow ligand exchange rates (on the chemical shift time-scale), and characteristic chemical shifts of bound ligands arising from the porphyrin ring current.
2.5.1 Rh / Sn isonicotinic acid system
As a control experiment to verify that carboxylic acids are neither bound to 118 nor result in its protonation or decomposition, 118·MeOH was titrated with benzoic acid in CDCl3. Small chemical shift changes (~0.1 ppm) of the benzoic acid aryl protons were observed during this titration, with H2 undergoing the most prominent shift. No significant changes to the porphyrinic resonances were observed. These results may imply weak coordination of the benzoic acid to the porphyrin, or may simply arise due to concentration dependence of the carboxylic acid chemical shifts due to self association.
Isonicotinic acid was chosen as the simplest ligand possessing both a pyridyl group and a carboxylic acid for coordination to Rh(III) and Sn(IV) respectively. The 1,4 disposition of these functionalities was selected to avoid a steric clash between simultaneously bound porphyrins. The portionwise addition of a suspension of isonicotinic acid to a solution of 118 in CDCl3 was monitored by 1H NMR spectroscopy. This afforded a 1:1 complex, 141, in which it is proposed that the ligand is bound to Rh via the nitrogen atom. The chemical shifts for this species are given in figure 2.28.
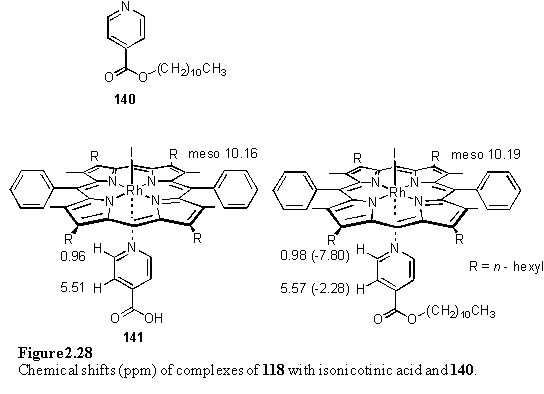
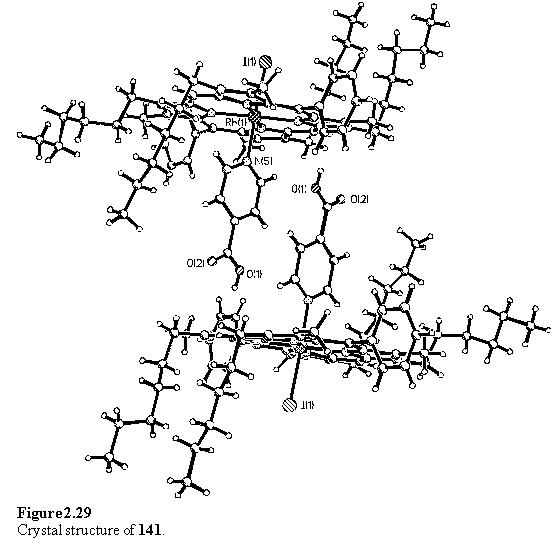
Free isonicotinic acid was not observed in the NMR spectrum due to its insolubility in CDCl3. Crystallization of 141 from a non-hydrogen bonding solvent was expected to afford a carboxylic acid dimer,291 as had been observed for a Ru(II) tetraarylporphyrin isonicotinic acid complex.292 However the crystals obtained from a DCM/hexane mixture did not display this hydrogen bonding motif, but instead consisted of a collapsed structure in which a pair of isonicotinic acid ligands lie parallel and offset to each other and the acidic proton make a close approach to a pyrrole ring (figure 2.29). The carboxylic acid proton is not disordered over the two oxygen atoms, as evidenced by the C-O bond lengths of 1.315(8) and 1.202(7) Å consistent with a single and a double bond.293 The distance between the pair of isonicotinic acid groups is 3.5 Å. No solvent molecules were located in the crystals and the porphyrins are almost exactly planar, with as rms atomic deviation of 0.07 Å from the least squares best fit plane. The porphyrin plane makes an angle of 86.18(8)° to the aryl group of the ligand. The driving force which favours this structure in preference to the hydrogen bonded acid dimer may be the requirement that the molecules pack densely in the crystal without leaving voids. Instead of the tilted structures with included solvent described in the previous sections, this molecule may pack efficiently by foregoing the strong hydrogen bonds between acids, and adopting the observed slipped structure with a weaker interaction between the acid and the porphyrin p system.
In order to determine an estimate of the complexation induced shifts for isonicotinic acid bound to 118, the isonicotinic acid ester 140 can be used as a model. Unlike isonicotinic acid, the NMR spectrum of 140 can readily be measured in CDCl3 and the spectrum of this compound cannot be complicated by association or zwitterion formation. Chemical shifts and Dd for the complex of 140 with 118 are given in figure 2.28.
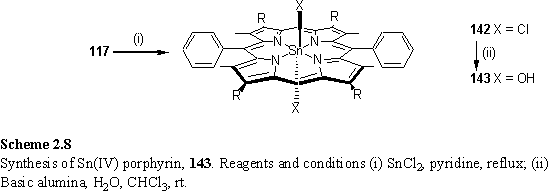
Metallation of 117 with SnCl2 afforded the dichloro Sn(IV) porphyrin 142, which was hydrolysed on wet alumina to dihydroxy Sn(IV) porphyrin, 143 (scheme 2.8).294 The hydroxyl resonance of 143 appears as a singlet peak at -7.83 ppm with satellites due to coupling to I = ½ Sn isotopes.
The reaction of 143 with isonicotinic acid was monitored by 1H NMR spectroscopy (figure 2.30). Addition of a suspension of isonicotinic acid to 143 in CDCl3 resulted in formation of a 1:1 complex, 144, within minutes. The Sn(OH) resonance broadened after addition of the acid, but sharpened with time as the acid complexed to the porphyrin. On standing overnight at room temperature 144 equilibrated to a mixture of 143, 144 and the 2:1 complex 145. Further addition of isonicotinic acid, allowing time for equilibration to occur, gave a solution in which 145 was almost the exclusive species. This kinetic preference for formation of a 1:1 Sn(IV) carboxylate complex, followed by equilibration has been previously observed for other carboxylic acids.289,295
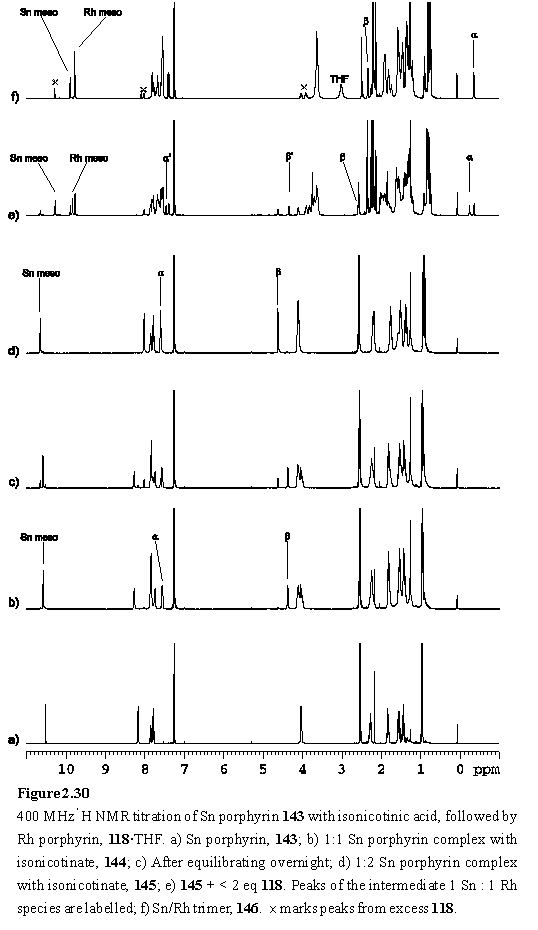
Selected chemical shifts of 144 and 145 are given in figure 2.31. A definitive assignment of the Ha resonance of 144, which lies close to the other aromatic resonances, was made with the assistance of a COSY spectrum. The spectrum of 145 was assigned by analogy with 144.
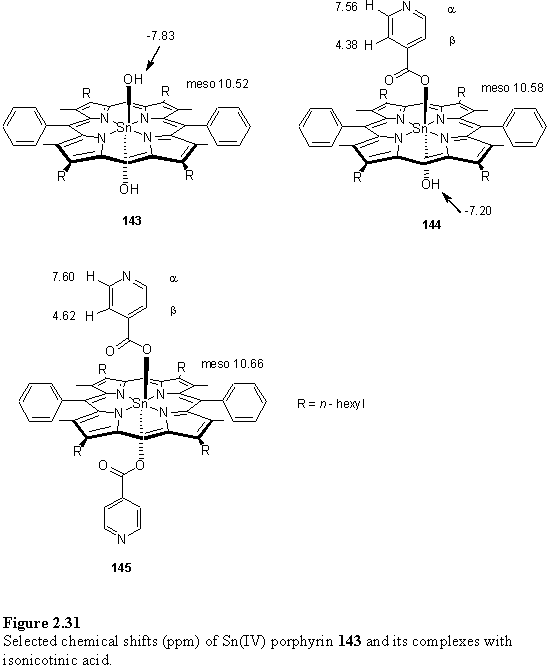
Addition of ~0.9 eq solid 118, as the THF solvate, to the sample of 145 resulted in an increase in the complexity of the NMR spectrum (figure 2.30 e) due to the presence of three species, in addition to THF displaced from 118. These are 145, the 1:1 complex formed between 145 and 118, and the 1:2 complex, 146. A COSY spectrum was acquired at this point to correlate the a and b resonances of these complexes. The chemical shifts are shown in figure 2.32. Further addition of 118 simplified the spectrum consistent with conversion of all of 145 and the intermediate 1:1 complex to 146. The remaining major resonances can be assigned to an excess of 118 and THF.
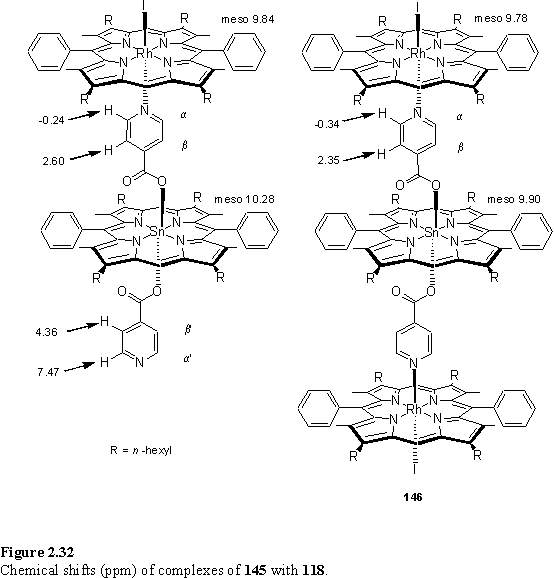
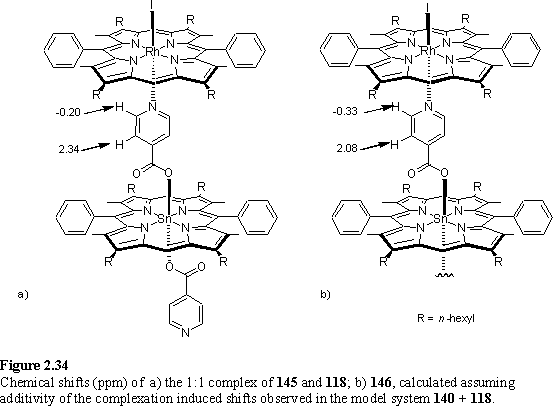
It is interesting to compare the observed chemical shifts of 146 with those predicted assuming an additivity of the complexation induced shifts (Dd) determined from the 1:1 complex. The agreement (figure 2.33) is remarkably good. However if the calculation is repeated (figure 2.34) using those Dd values obtained from the model system 140 + 118 whilst the agreement is reasonable for Ha it is poor for Hb with a discrepancy of ~0.25 ppm between calculated and experimental chemical shifts. An explanation for this is that coordination of the Rh porphyrin may sterically restrict the conformations available to the isonicotinate group, and thus the average shielding experienced by Hb as a result of the Sn porphyrin is altered as the Rh porphyrin binds. Thus the complexation induced shift of these protons is not comparable to the model system. The Ha protons are further from the Sn porphyrin and the chemical shift is less sensitive296 to the conformation of the isonicotinate group so undergoes the same complexation induced shift observed in the model system upon binding of 118.
Crystals were grown from the NMR sample (redissolved in DCM and layered with MeOH) and X-ray analysis revealed the structure of 146 (figure 2.35) corroborating the NMR evidence for the existence of this molecule, and clearly demonstrating the potential of this chemistry to specifically assemble a large structure from simple components. The two rhodium porphyrins are equivalent by symmetry and almost exactly planar with an rms deviation from the best fit plane of 0.06 Å. The whole isonicotinate moiety is also planar (rms deviation from planarity of 0.03 Å), and perpendicular to the Rh porphyrin with an angle of 89.97(6)° between the best fit planes. The Sn porphyrin is slightly tilted with respect to the Rh porphyrins, with an angle of 8.64(5)° between the best fit planes. In the solid, the molecules are interdigitated such that the volume between the stack of porphyrins is filled by a pair of neighbouring molecules.
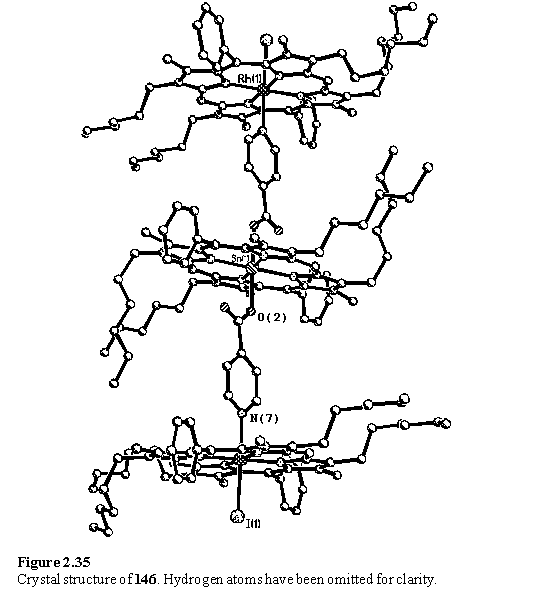
To ensure that the crystal picked for structure determination was not a minor component of the isolated crystals, these were redissolved and an NMR spectrum acquired. The majority of peaks observed in the spectrum could be assigned to 146 indicating that the crystal examined by diffraction was indeed representative of the bulk sample.
2.5.2 Porphyrins functionalized with pyridyl and carboxylate as ligands
To extend this self assembly strategy to prepare arrays of porphyrins the isonicotinic acid may be replaced with a porphyrin based building block. There is some precedent138 for the use of carboxylic acid substituted porphyrins as ligands for Sn(IV) porphyrins as was discussed in the previous chapter.
The most straightforward such ligand is 148 and this was prepared via methyl ester 147. 147 was synthesized in a statistical condensation of 4-formyl pyridine and 4-formyl methyl benzoate with 116 after deprotection and decarboxylation using the standard procedure, followed by DDQ oxidation. 147 was separated from the mixture of three porphyrins by chromatography prior to ester hydrolysis with KOH in wet THF. A red precipitate formed during this reaction, presumably the potassium salt of 148. This could be solubilized by treatment with HCl(aq) and extraction of the green porphyrin dication into CHCl3. Washing with water effected deprotonation of this cation to restore a red colour to the solution. The product was chromatographed on silica, and the purity judged to be satisfactory by NMR although the compound could not be obtained analytically pure. As had been feared, 148 was almost insoluble in all common pure solvents. However the solubility in CHCl3 was considerably enhanced by addition of methanol. The NMR spectrum of 148 in a CDCl3 + CD3OD mixture showed no evidence of zwitterion formation as judged by the similarity of the chemical shifts of the pyridyl Ha resonances of 147 and 148, which appeared at 9.02 and 8.90 ppm respectively.
To assemble the multiporphyrin structure, weighed amounts of solid 148 and 143 in a 2:1 molar ratio were mixed and CDCl3 added. Initially the mixture failed to dissolve but dissolution was assisted by sonication and warming. After 2 days the NMR spectrum was acquired and displayed a pair of characteristic doublets at 7.03 and 5.19 ppm corresponding to the bound aryl carboxylate group of 149. Disappointingly the meso region of the spectrum was not as clean has had been hoped for, although the two major peaks could be assigned to the meso protons of the tin and free-base porphyrin components. The proposed spectral assignments are given in figure 2.36 b.
Addition of two equivalents of solid 118·THF afforded the porphyrin pentamer 150. Although the 1H NMR spectrum (figure 2.37) of this was not totally clean the major peaks could be assigned to 150. Small doublets around the more intense resonances of the bound carboxylate suggest that these impurities are due to unexpected tin porphyrin containing products, perhaps resulting from the presence of unintended oxygen donors which coordinate to the tin.
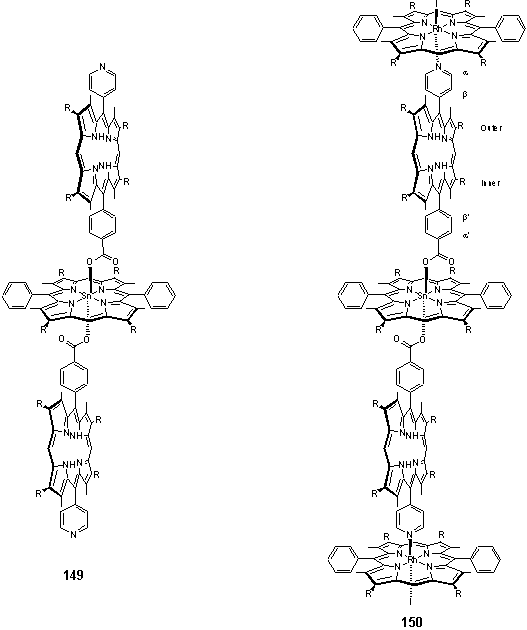
The free-base porphyrin pyridyl Hb resonance is clearly visible at 5.70 ppm and the Ha peak can be identified at 1.17 ppm using a COSY spectrum. An NOE links this peak to the free-base b-methyl at 0.73 ppm. The aryl carboxylate group appears as a pair of doublets at 6.86 and 5.08 ppm and these show an NOE to the other b-methyl at 1.64 ppm.
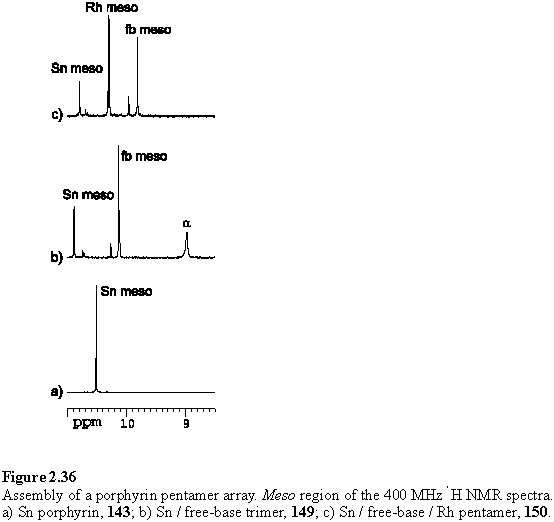
Of the three main meso resonances the Sn porphyrin can be identified at 10.80 ppm by integration. The free-base porphyrin meso appears at 9.81 ppm as this proton lies within the shielding region of both the Sn and Rh porphyrins. This assignment is also consistent with the NOESY connectivity, with crosspeaks connecting the resonances in the sequence: meso - hexyl H1 - b-Me - aryl. The Sn and Rh porphyrin hexyl H1 and b-methyl groups can similarly be identified by the NOESY connectivity from the meso proton. The assignments are given in figure 2.37. Due to congestion of peaks around 1.5 ppm it has not been possible to assign all of the hexyl resonances from the COSY spectrum. A peak at 3.63 ppm was assigned to THF, displaced from 118, and disappeared after the sample was dried in vacuo and redissolved in CDCl3. It is this spectrum which is displayed in figure 2.37.
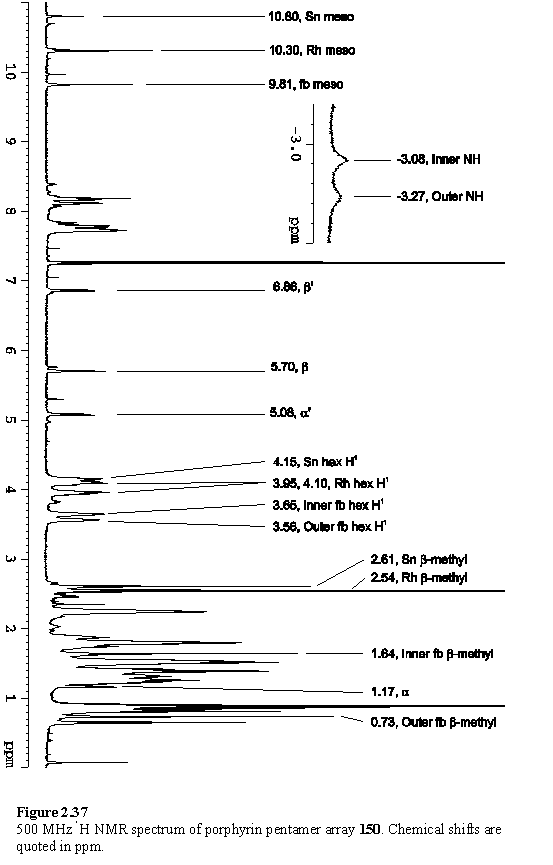
The free-base porphyrin component has two degenerate tautomers in which the pyrrolic protons are inequivalent. The rate of exchange of these protons by tautomerization is slow on the chemical shift time-scale and a pair of broadened singlet peaks at -3.08 and -3.27 ppm results.
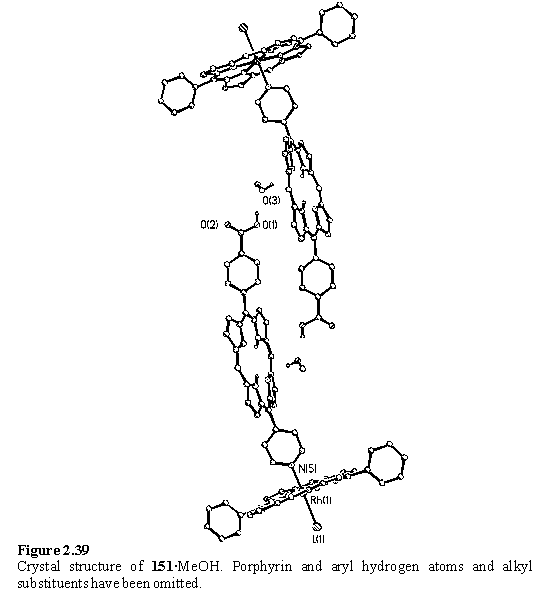
Attempts were made to obtain X-ray quality crystals of 150 using the same layered DCM/methanol technique which had proved successful for 146. Black crystals were obtained but these were found to be the Rh - free-base porphyrin dimer, 151, with no Sn porphyrin present.
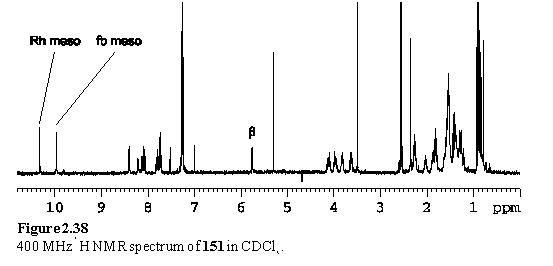
The 1H NMR spectrum of the crystals (figure 2.38) suggested them to be largely composed of 151. The NMR spectrum of the mother liquor displayed four meso resonances (10.65, 10.52, 10.30, 9.81 ppm) none of which appeared at the same chemical shift as the Sn porphyrin meso of 150. However doublets at 6.79 and 4.85 ppm demonstrate the presence of remaining Sn porphyrin bound carboxylate, but that this species is not the same as 150. A doublet at 5.70 ppm arises from the pyridyl Hb of a species derived from 148 coordinated to a Rh porphyrin. During the time before crystallization the carboxylic acid appears to have been at least partially displaced from the Sn porphyrin to yield 151 and the other unknown products remained in the mother liquor. The large excess of methanol competing with the acid for binding to Sn may have been responsible for this.
The structure of 151 contains a hydrogen bonded network between the carboxylic acid group, a methanol molecule and the pyrrolic nitrogen atom of a neighbouring molecule. This hydrogen bonding organizes the molecules of 151 into dimeric units (figure 2.39). This type of hydrogen bonding to the core of free-base porphyrins has been reported230, 297,298 in the literature and will be encountered again in section 3.3.1.