3.3 Porphyrins for SAMs
In order to be able to directly functionalize gold surfaces with metalloporphyrins a versatile synthetic route to pure porphyrin with thiol or disulfide functionality was required. Tour et al. reported370 in situ cleavage of aryl thioacetates on a gold surface to form SAMs, but at the commencement of the work described in the following sections this had not been generalized to alkyl thioacetates, although subsequently Lindsey et al. have demonstrated cleavage and adsorption of porphyrins functionalized with benzylic thioacetates.172
Porphyrin alkyl thiols of the type in chart 3.1 were selected as synthetic targets. As discussed in section 1.7, the presence of one or two thiol groups has been reported to produce monolayers in which the porphyrin is oriented perpendicular or parallel to the gold surface respectively.174 Ether linkages between the aryl groups of the porphyrin and the alkyl chains were chosen due to their synthetic expediency, hydrolytic stability and absence of hydrogen bond donor ability. The unsubstituted meso positions permit oxidation to a nitro or oxo porphyrin with concomitant increase in the affinity of the Zn metallated derivative for axial coordination of nitrogenous bases.371 The alkyl substituents at the b positions enhance the solubility of the porphyrin in organic solvents. Alkyl chains packing underneath a large head group in a monolayer have been reported to be responsible for enhanced stability of the monolayer.372
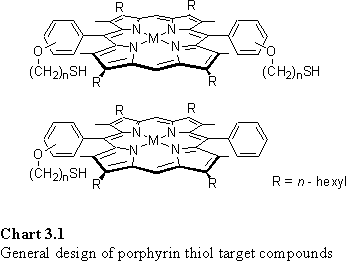
The syntheses of these metalloporphyrins will be discussed in the following sections.
3.3.1 From phenolic porphyrins
The initial synthetic effort focused on the alkylation of phenolic porphyrins with alkyl bromides according to scheme 3.12. The phenolic porphyrin 196 was prepared by the standard porphyrin synthesis procedure, without protection of the hydroxyl groups of 3-hydroxybenzaldehyde. The monophenol porphyrin 197 was prepared similarly in a statistical condensation of dipyrromethane with 3-hydroxybenzaldehyde and benzaldehyde. The large difference in polarity between the three products facilitated chromatographic separation. 3-Hydroxybenzaldehyde was selected in preference to the isomeric 2-hydroxybenzaldehyde as this was expected to give rise to atropisomeric products due to restricted rotation about the meso-Ar bond. Aryl porphyrins with 4-hydroxy substituents have been reported to suffer from some instability, especially under basic conditions, with respect to air oxidation to a quinone type structure.373
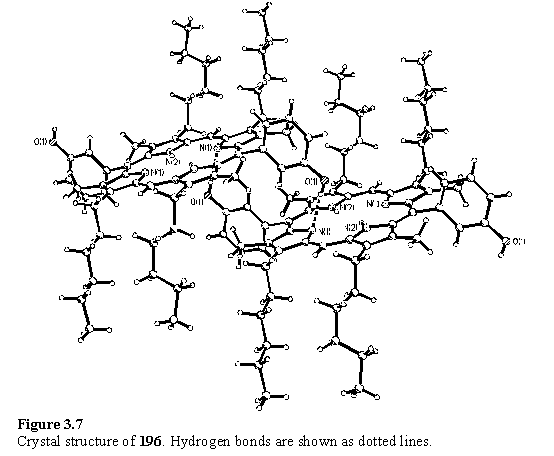
X-ray structures of 196 and 197 were obtained and a packing plot is shown in figure 3.7. The packing of 196 takes the form of an infinite tape of molecules in which each hydroxyl proton is hydrogen bonded to the pyrrolic nitrogen atom of an adjacent molecule. Each molecule forms a total of four hydrogen bonds. 197 adopts an almost identical structure, except the hydroxyl group is disordered with equal occupancy over the two sites that are fully populated in 196. This model of the solid state structure may imply that the hydrogen bonding between the porphyrins is not cooperative as there appears to be no preference for formation of hydrogen bonded dimers, or an infinite tape in which all the hydrogen bonds lie in the same direction along the tape. However on the basis of the X-ray data alone it is impossible to rule out a fully ordered structure within each of the tapes, but disorder between adjacent tapes.
195 was prepared by statistical reaction of 1,11-dibromoundecane with potassium thioacetate.337 The literature describes alkylation of phenolic porphyrins with K2CO3 in DMF374,375 and Cs2CO3 in DMF.376 Reaction of 195 with 197 under the Cs2CO3 conditions yielded a mixture from which the product 215 was isolated after preparative plate chromatography and zinc metallation. The product was zinc metallated due to the problem of partial metallation occurring during the chromatography as a result of the presence of Zn sulfide fluorescer in the plates.377
Under the same alkylation conditions 196 afforded a diverse mixture of products from which the diacylated compound (scheme 3.12) was isolated and identified from the 6H CH3CO2 singlet at 2.37 ppm in the 1H NMR spectrum and from a molecular ion at m/z 1034 in the FAB mass spectrum. Evidently a significant side reaction is transesterification of the thioester of 195 with the phenol of 196 to yield a phenol ether and a thiol. Either alkylation or acylation can occur at each phenol group. If alkylation occurs then the other terminus of the alkyl chain may be functionalized with either a thioacetate or a thiol. Therefore it is unsurprising that a large number of products was observed. To avoid this difficulty 196 was alkylated with 11-bromoundecanol and zinc metallated, with a view to converting the alcohol moiety to thiol via a tosylate and thioacetate (scheme 3.13). The 1H NMR spectrum of the alkylated product was more complex than had been expected, with a meso resonance split into two signals in a 1:1 ratio. This splitting of lines is most likely due to the presence of cis and trans atropisomers which interconvert at a rate which is slow on the NMR chemical shift time-scale. The CH2OH group appeared as an upfield shifted multiplet which could best be interpreted as a pair of overlapping quartets at 2.91 and 2.94 ppm with J = 6 Hz. In dilute samples a 2H multiplet, believed to be composed of two overlapping triplets with J = 5 Hz was observed at 0.38 ppm. In other samples this signal was broadened, and was assigned to the hydroxyl protons, with the multiplet structure arising from coupling to the CH2 group. Coupling to hydroxyl protons is observed in dilute and acid free samples due to slow exchange of these protons with each other and water.378 The upfield shifting of the CH2OH resonance from its usual position at around 3.6 ppm is consistent with coordination of the alcohol to the Zn ion of the porphyrin. Bound and unbound hydroxyl groups are in fast exchange giving rise to a single resonance, but this is split into two by the existence of atropisomers.
The ditosylation of this diol did not afford clean product (by 1H NMR), and at this point < 10 mg of material was available. This reaction scheme suffers the drawback that each reaction must occur at two sites, so any side reactions will lead to poor yield and a multitude of products which must be separated. Therefore this approach was abandoned in favour of an alternative synthesis of porphyrins using aldehydes already functionalized with protected thiols.
3.3.2 Porphyrin synthesis with functionalized aldehydes
The synthesis of porphyrins by BF3 catalysed condensation of pyrrole with an aldehyde derived from alkylated 4-hydroxybenzaldehyde carrying a terminal thioacetate group was reported by Shimazu,177 although the synthesis of the aldehyde was not described. It was decided to adopt the aldehydes of this type in the current work, and to develop appropriate syntheses, according to scheme 3.14.

The alkylation of hydroxybenzaldehydes with bromoundecanol utilized an excess of the aldehyde to ensure complete consumption of the bromide. Owing to the water solubility of the hydroxybenzaldehydes in both their neutral and deprotonated forms, and the insolubility of the alkylation products 198, 204 and 207 in water, the excess starting material could be removed by recrystallization from an ethanol/water mixture, or alternatively a basic wash during the work-up. The crude products were often obtained in satisfactory purity in yields of 80 % to almost quantitative, although further purification could be achieved if required by recrystallization. Batches of 198 and 204 in greater than 5 g quantities could be prepared using this method.
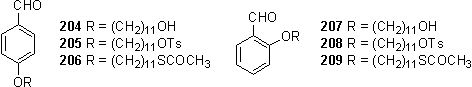
Tosylation of the alcohols was carried out by reaction with TsCl in DCM with Et3N as base, warming from 0 °C to room temperature. Best results were obtained using concentrated solutions of TsCl, up to 0.8 M, with extra portions of Et3N and TsCl being added to force the reaction to completion. Product purification required column chromatography, although after optimization of the solvent system this was readily applicable to separation of 5 - 10 g quantities of the tosylates which were typically isolated in greater than 80 % yield.
Conversion of tosylates to thioacetates was achieved by reaction with potassium thioacetate in PEG 400 according to the literature.337 200 and 206 were conveniently purified by recrystallization and isolated in quantities exceeding 5 g in yields of greater than 80 %. Purification of the 2-substituted aldehydes 207 and 209 was more problematic as these were found to be less amenable to crystallization thus occasionally necessitating the use of column chromatography.
The analogue, 203, with a 12 carbon alkyl chain was prepared by the same route, although on a smaller scale.
Despite fears about the lability of the thioacetate protecting group to TFA, the aldehydes were successfully condensed with dipyrromethane with TFA catalyst, followed by DDQ oxidation, to afford porphyrins 210, 211, 212, 217 and 218. Dry THF was chosen as the reaction solvent in preference to MeOH to avoid acid catalysed transesterification of the thioester. Unsymmetrical porphyrins were prepared by a statistical reaction of dipyrromethane with a mixture of benzaldehyde and substituted aldehyde. Atropisomers of 218 could be detected by silica gel tlc although it was not possible to cleanly separate these on a preparative scale.
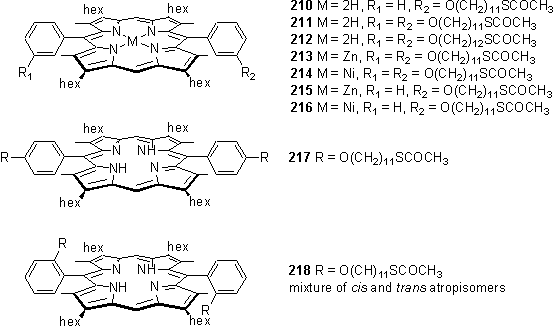
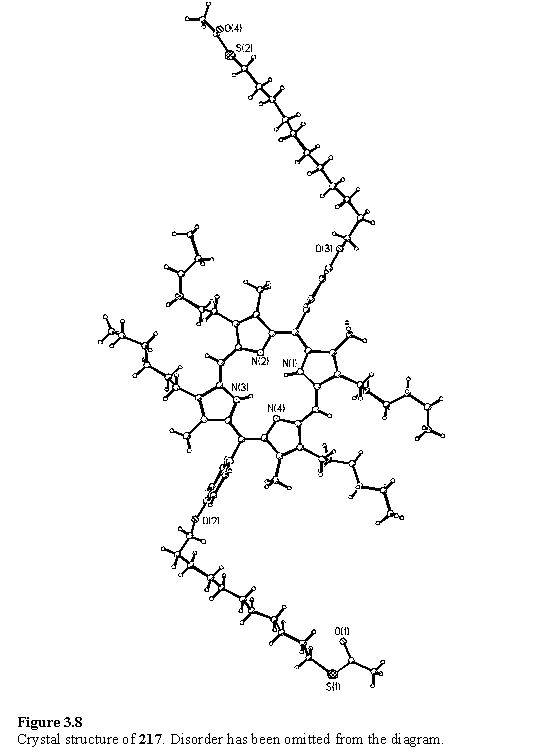
The thioacetate group was not deprotected to a significant extent under the porphyrin synthesis conditions as evidenced by the observation of the CH2SCOCH3 resonance at 2.8 ppm and the COCH3 group at 2.3 ppm in all the products. The symmetrical porphyrins were isolated in quantities up to 2 g with yields in the range 50 - 70 %. Remarkably, diffraction quality crystals of 217 were obtained which confirmed the molecular structure (figure 3.8). The structure is badly disordered due to the large conformational flexibility of the alkyl groups of 217.
In order to assess the necessity of the b-hexyl groups for solubility of the porphyrins, and the effect of the b substituents on self assembled monolayer structure, for comparison a derivative, 220, lacking these substituents was prepared according to scheme 3.15. The dipyrromethane, 219, was prepared by either the method of Lindsey,379 although the yields claimed could not be reproduced, or by the method of Anderson380 in which paraformaldehyde is thermally cracked prior to reaction with pyrrole. The 13 % yield of 220 is substantially lower than that obtained for the b substituted 211 (up to 70 %), although an attempt at synthesizing 220 under the TFA catalysed conditions used to prepare 211 did not yield any porphyrinic products as judged by tlc.
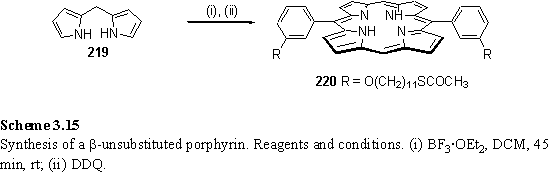
3.3.3 Metallation reactions
Zinc metallation of 210 and 211 was performed by warming a chloroform solution of the porphyrin with Zn(OAc)2 and several drops of methanol to assist in solubilizing the Zn salt. The reaction was always found to proceed to completion as observed by a single clean meso resonance in the 1H NMR spectrum and the disappearance of the pyrrole NH resonance at -2.4 ppm.
Nickel metallation required the more forcing conditions of refluxing a chloroform/methanol solution of porphyrin with Ni(OAc)2·4H2O for several hours. The reaction could be monitored by silica gel tlc, with the Ni metallated product appearing as a red non-fluorescent spot at a higher Rf than the red-brown and fluorescent starting material.
Rhodium metallation of 217 was carried by reaction with Rh2(CO)4Cl2 followed by iodine oxidation under the same conditions used to prepare 118. This porphyrin was chosen to test the metallation reaction as it is symmetrical and cannot exhibit atropisomerism, unlike 211, which serves to complicate the 1H NMR spectrum by doubling resonances. A successful Rh metallation of 211 could lead to 3 atropisomeric products as the phenyl ethers can adopt a cis or trans arrangement with respect to each other, and in the trans form the iodo group could also be cis or trans with respect to the ethers.
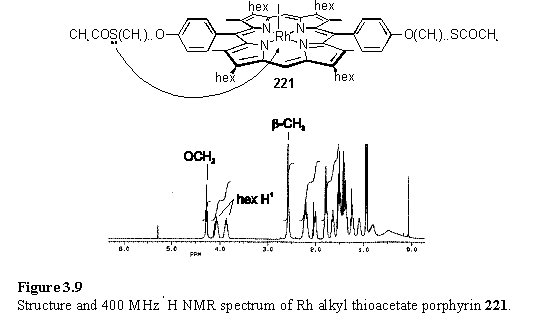
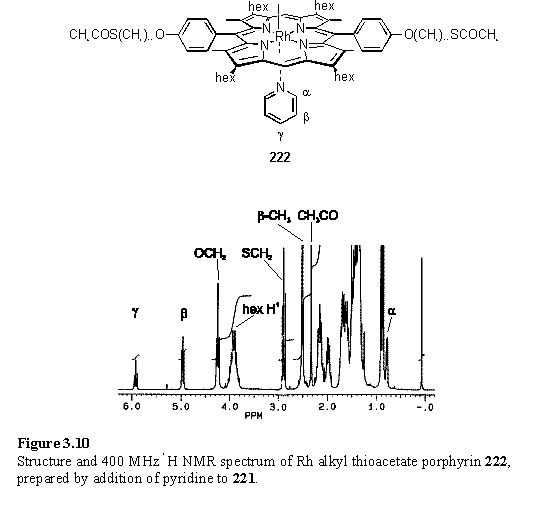
The product, 221, did not show the expected CH2S and COCH3 resonances in the NMR spectrum, but instead showed broadened and upfield shifted resonances attributed to the C11 alkyl chain (figure 3.9). Elemental analysis and mass spectral data were consistent with the proposed structure. The cause of broadness in the 1H NMR spectrum is believed to be exchange between thioacetate group coordinated to the Rh centre and free thioacetate as was discussed in section 2.8. Infrared spectroscopy supports this conclusion and two carbonyl stretching bands were observed in the spectrum of 221, one at 1733 cm-1 corresponding to coordinated thioacetate, and the other at the more usual frequency of 1684 cm-1 corresponding to unbound thioacetate. This band was observed at 1686 cm-1 in the parent compound 217. A shift to higher frequency on coordination is consistent with donation of the sulfur lone pair to Rh, which decreases the contribution of the resonance form with a C-O single bond to the electronic structure of the thioacetate thus increasing the double bond character. Addition of pyridine to 221, followed by drying afforded 222. The 1H NMR spectrum of 222 (figure 3.10) is dramatically different from 221 in that the alkyl resonances are sharp and with chemical shifts typical of these substituted porphyrins. The shielded resonances of coordinated pyridine are visible at 0.78 (Ha), 4.96 (Hb) and 5.92 (Hg) ppm which are almost identical chemical shifts to the pyridyl resonances of 124. The thioacetate group has reappeared at 2.89 (CH2S) and 2.33 (CH3) ppm and the IR spectrum shows a single carbonyl stretching band at 1686 cm-1. These results are also consistent with a thioacetate group which coordinates to Rh in 221 but is displaced by pyridine on formation of 222. As thiol ligands were found to bind to rhodium porphyrin more strongly than thioacetates, if the thioesters of 221 were to be deprotected then a single thiol group would almost certainly coordinate to the rhodium centre.
3.3.4 Cleavage of thioacetate
A variety of conditions were explored for cleavage of the thioacetate protecting group. Due to the air sensitivity of the thiol product all reactions were freeze-thaw degassed and carried out under an inert atmosphere. Oxidation of porphyrin dithiols is especially deleterious due to formation of insoluble polymers.381 The reducing conditions of NaSMe in MeOH used for deprotection of 183 could not be applied to these porphyrins due to poor solubility of the porphyrin in MeOH. Therefore other conditions were sought in which a thiolate could be generated in a solvent in which the porphyrin dissolves. Mercaptoethanol is miscible with common organic solvents and 210 on refluxing with mercaptoethanol in THF with Et3N as a base for 12 h yielded thiol porphyrin 227. The mercaptoethanol is significantly more polar than the porphyrin and could be retained on the silica gel during chromatographic purification. Use of the stronger organic base DBU in refluxing wet THF, with mercaptoethanol reduced the reaction time to 90 minutes.
For larger scale deprotections, up to gram scale, an alternative procedure, modified from the literature,382 using aminolysis with butylamine in refluxing THF was adopted. DTT was the preferred reducing agent as this showed superior retention on silica than mercaptoethanol, and also offers greater protection from oxidation due to the formation of a disulfide in a six membered ring in its oxidized form.383
The success of the deprotection could be assessed by 1H NMR spectroscopy. The outcome of a successful reaction was a quartet at ~2.5 ppm assigned to CH2SH and the disappearance of the thioester resonances of the starting material. Disulfide contaminants were indicated by a triplet at ~2.6 ppm. The CH2S resonance of Zn porphyrin 224 was observed at 2.38 ppm, whereas that of the corresponding free-base porphyrin 223 occurred at 2.47 ppm. The SH protons of 224 were identified from the COSY spectrum as a triplet at 1.17 ppm with 3J = 8 Hz. The slightly upfield shifted CH2S resonance of 224 may arise from weak coordination of the thiol to the zinc centre. Other researchers have also reported little affinity of zinc porphyrin for thiol.171
3.3.5 Oxidative instability of thiol porphyrins
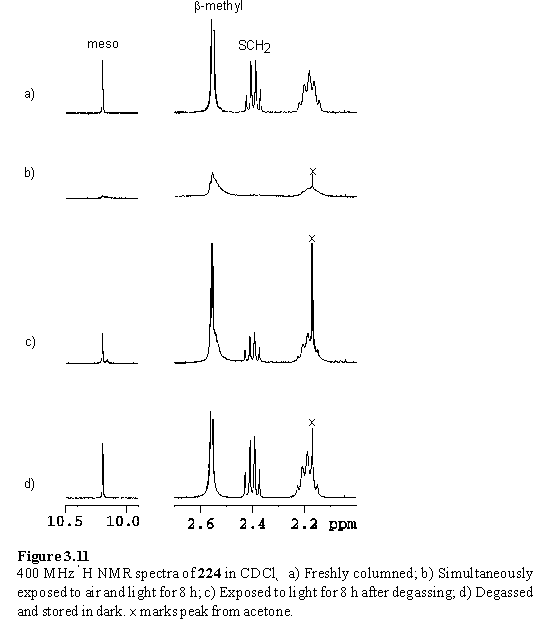
During purification of the thiol porphyrins it was observed by visual inspection, tlc and NMR that after chromatography the compounds rapidly became impure if left in solution. This problem was found to be especially acute with the Zn dithiol compound 224 and was believed to be due to photosensitized oxidative polymerization of the thiols to disulfide by singlet oxygen.384 Ready oxidation of thiol porphyrins during work-up has been reported in the literature.385,386 To verify this hypothesis identical samples of 224 in CDCl3 were prepared, and one was exposed to ambient light for one day, whereas light was excluded from the other. The sample exposed to light developed a brown precipitate, but the sample that had been stored in the dark was unchanged both by visual inspection and in its 1H NMR spectrum. The necessity of oxygen was confirmed by division of a chloroform solution of 224 amongst 3 Schlenk tubes, two of which were degassed. One of the degassed samples was exposed to light, whilst light was excluded from the other. The third control sample was exposed to air and light, and developed a brown colour within 4 hours, and a precipitate after 8 hours. After this time the solvent was evaporated and the samples were prepared for NMR, and stored in the dark until acquisition of spectra. The 1H NMR spectrum of the sample that had been simultaneously exposed to air and light showed many broad resonances indicative of extensive decomposition. The spectrum of the degassed sample that had been exposed to light showed little change, with a clearly visible CH2SH quartet. Evidence for a minor decomposition product, probably arising from incomplete degassing, was a small additional meso peak. The sample that had always been excluded from light had an unchanged 1H NMR spectrum (figure 3.11).
As the products of the oxidation reaction of 224 came out of solution it was not possible to observe formation of disulfide by 1H NMR. Model compound 231 was dissolved in CDCl3 and dodecanethiol added. Otherwise identical, non degassed, samples were stored for 2 days in the presence and absence of light, after which time 1H NMR spectra were recorded. Only the sample which had been exposed to light showed the appearance of a disulfide CH2S triplet at 2.68 ppm.
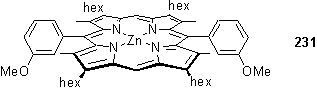
Acting upon these observations, thiol substituted porphyrins were stored in sealed nitrogen filled vials in a freezer at -20 °C. Solutions were handled rapidly under reduced light, and where possible under an inert atmosphere.
3.3.6 Macrocyclic disulfide substituted porphyrins for SAMs
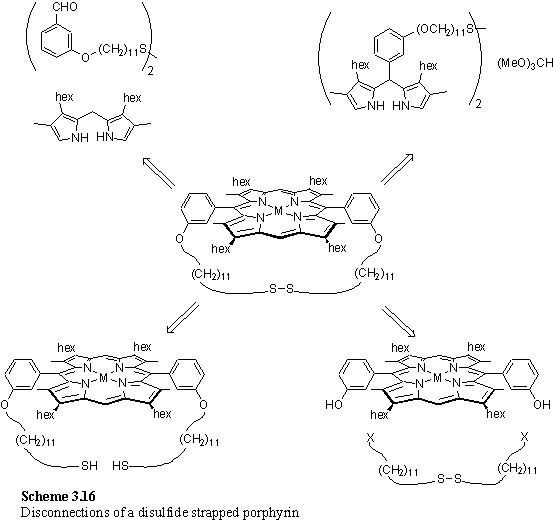
As discussed above, dithiol porphyrins suffered from the disadvantage of air sensitivity, and oxidized rapidly when exposed to light to form insoluble material. An additional, but unrelated, problem was the inseparability of the atropisomers of 218. A mixture of atropisomers was considered undesirable for SAM formation as each atropisomer has a quite different geometry and adsorption of both on a gold surface would lead to a heterogeneous SAM. All of the dithiol porphyrins need only adsorb through a single thiol group, leaving the other free, as is believed to be the structure of linear alkane dithiols such as 1,8-octanedithiol.387 A solution to both of these problems seemed to be the use of macrocyclic disulfides instead of dithiols. The strapping of the phenol ether groups was expected to force adoption of exclusively the cis atropisomer and by necessity on adsorption on a gold surface both sulfur atoms would simultaneously contact the surface and become bound. In addition the disulfide lacks the ability to become oxidized to polymeric material.
Two approaches were used to synthesize these macrocycles, direct oxidation of a dithiol, and condensation of dipyrromethane with a dialdehyde.388 Alternative syntheses that were not attempted are dialkylation of a phenolic porphyrin with the strap fragment,389 and condensation of a strapped dipyrromethane with trimethyl orthoformate.390 The four disconnections are shown in scheme 3.16.
3.3.7 Preparation of macrocyclic disulfides from dialdehydes
Dialdehydes 232 and 233 were synthesized by cleavage of the thioester groups of 200 and 209 followed by oxidation to a disulfide (scheme 3.17). The CH2S resonances of these compounds are triplets at 2.67 and 2.68 ppm respectively, which are typical values for alkyl disulfides.391
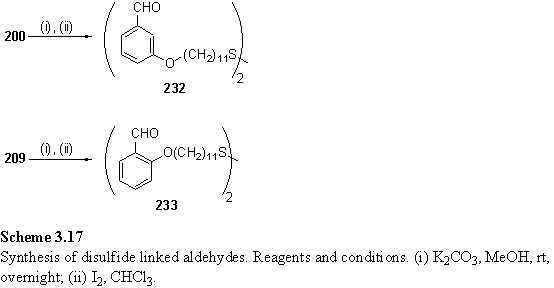
Dialdehyde was condensed with dipyrromethane (scheme 3.18) under a modification of the standard porphyrin synthesis conditions in which the concentration of aldehyde was reduced by approximately twenty-fold from ~65 mM (for synthesis of 211) to 2.8 mM. Oxidation of the porphyrinogen intermediate with DDQ afforded the product, 234 in a yield of 53 %.
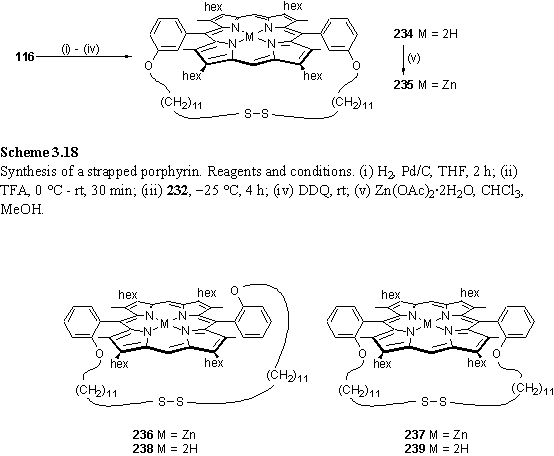
Evidence for the proposed cyclic disulfide structure is found in the 1H NMR spectrum. The CH2S resonance is not present at 2.7 ppm, but is shielded and overlaps with the hexyl H2 resonance at 2.21 ppm as judged from the 12H integral of this multiplet (figure 3.12 a). The strap constrains the disulfide in the shielding region over the porphyrin plane, as has been observed in other strapped and ‘basket handle’ porphyrins.388-390,392,393 The high resolution ES MS spectrum gave a molecular ion with m/z 1257.8930 compared to a calculated mass for [M+H]+ of 1257.8925 also confirming the identity of the product.
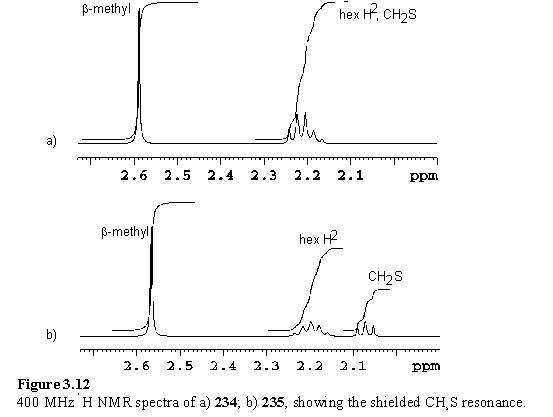
The shielded CH2S triplet is clearly observed at 2.07 ppm in the zinc derivative 235 as it does not overlap with the hexyl H2 multiplet (figure 3.12 b). The hexyl H1 resonances of 234 and 235 do not appear as simple triplets as the CH2 protons are rendered inequivalent by the strap which passes over one face of the porphyrin. A single meso resonance was observed, suggestive of a single atropisomer or rapid atropisomerization on the NMR time-scale.
Condensation of 233 with dipyrromethane gave a mixture of two major products which after zinc metallation were separated with difficulty by preparative thin layer chromatography. These products are proposed to be a pair of trans and cis atropisomers, 236 and 237 respectively. Inspection of a CPK model of the trans isomer revealed the strap to be long enough to span the aryl groups around the periphery of the porphyrin without considerable distortion of the porphyrin, but with loss of conformational freedom of the strap. The products were identified by their 1H NMR spectra. The compound assigned to the cis isomer showed a shielded CH2S triplet at 2.48 ppm, whereas the trans isomer displayed a deshielded triplet at 2.88 ppm consistent with the CPK model which requires the central region of the strap to be located around the porphyrin periphery. The dispersion of the alkyl chain resonances is greater in 236 as the strap is restrained to lie closer to the porphyrin. The strap of 235 is shielded to a greater extent than that of 237 as the meta substitution pattern restrains the strap to lie on average closer to porphyrin plane. MALDI TOF mass spectrometry gave peaks at m/z 1324 [M]+ and 1259 [M-Zn]+ for 236 and at 1321 and 1259 for 237 consistent within the experimental error of this instrument with the calculated molecular weight of 1321. No unusual features, such as a red shifted Soret band, were observed in the UV/visible spectra, suggesting that the porphyrins are not considerably, if at all, distorted from planarity by the presence of the strap.388 Despite repeated efforts, an X-ray structure of 236 could not be obtained, even with the use of synchrotron radiation on two promising batches of crystals.
236 and 237 could be demetallated by treatment with acid to form a green dication which was deprotonated with Et3N and washed with water to generate free-base porphyrins 238 and 239 respectively. The NMR spectrum of 238 indicated the presence of a small quantity of 239.
3.3.8 Preparation of macrocyclic disulfides by oxidation of dithiols
The reaction of 224 at a concentration of 40 mM in DCM with iodine, followed by remetallation with zinc, afforded 235 in a yield of 87 %. Material obtained in this way had an identical 1H NMR spectrum to that produced by the dialdehyde route.
Similarly a sample of 218 was deprotected to thiol by aminolysis with BuNH2 and treated with iodine in DCM, with a porphyrin concentration of 0.1 mM. The reaction did not proceed cleanly, probably due to a slow rate of cyclization of the trans atropisomer, which lead to formation of oligomeric products. However after zinc metallation and preparative tlc, 237 was isolated in 9 % yield and was also identical to material prepared by the alternative scheme. It is not clear whether the trans isomer was not formed during the oxidative cyclization or whether it was simply not isolated from the mixture of products.
3.3.9 Thermal atropisomerization
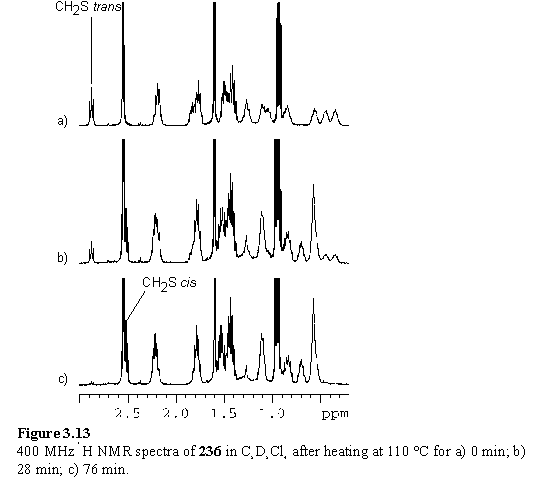
The thermal atropisomerization of 236 and 237 was investigated by NMR spectroscopy. In a preliminary experiment to determine the temperature required for isomerization, a sample of 236 in C2D2Cl4 was heated in the NMR spectrometer and 1H spectra recorded at regular temperature increments. At 107 °C after several minutes the resonances of 237 appeared. The temperature was increased to 117 °C and after a period of minutes the resonances of 236 were replaced by those of 237 exclusively, indicating that 236 has only kinetic stability. The spectrum of 237 was also acquired at 117 °C to confirm the identity of the reaction product.
The rates of isomerization of 236 and 238 in C2D2Cl4 at 110 °C were measured by immersion of NMR samples in a thermostatically controlled oil bath for a measured period of time, followed by quenching with an ice bath and acquisition of an 1H NMR spectrum (figure 3.13). The spectra were quantified by integration of the meso resonance which was normalized for each spectrum in the series, and the quantity of trans isomer determined from the CH2S integral. Data were fitted to a first order model to obtain rate constants (table 3.1). The data point to a more rapid isomerization of the free-base porphyrin 238 than the zinc porphyrin 236, consistent with what is already known about atropisomerization rates and ascribed to a rigidification of the porphyrin on metallation.394
|
236 |
238 |
|
0.037 (-0.998) |
0.10 (-0.983) |
|
0.039 (-0.997) |
0.12 (-0.994) |
Table 3.1
Rate constants (min-1) for trans to cis atropisomerization of strapped porphyrins in C2D2Cl4 at 110 °C. The correlation coefficient of the least squares fit is given in parentheses.
3.3.10 Surface bound multiporphyrin receptors
From CPK models it was predicted that a covalently linked porphyrin dimer of the form of 246 would have the correct spacing between the metal centres to bind a 4,4'-bipyridine guest. Although the dimer is flexible, the conformations accessible might be restricted if the receptor were tethered into a SAM surface. The diaryl acetylene structure can readily be assembled by the Pd catalysed chemistry302 used for synthesis of the dendrimer wedges of section 2.6.1. The central aryl group provides an attachment point for an alkyl thiol chain for binding to gold. In order to apply the chemistry discussed in section 3.3.2 for synthesis of the alkyl thiol group, 3,5-diiodophenol, 241, was prepared according to the literature from 3,5-dinitro anisole (scheme 3.19).395
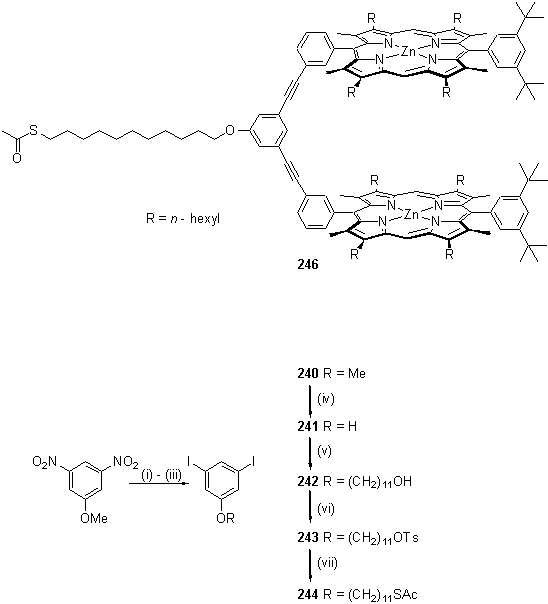
Scheme 3.19
Preparation of a diiodoaryl alkyl thioacetate. Reagents and conditions. (i) AcOH, PtO2, H2, 5 h; (ii) NaNO2, H2SO4, AcOH, 0 °C, 1 h; (iii) NaI, urea, I2, H2O, CHCl3, rt, 1 h; (iv) HI, AcOH, reflux; (v) Br(CH2)11OH, Cs2CO3, DMF, 2 d; (vi) TsCl, Et3N, DCM, 2 d; (vii) KSCOCH3, THF, MeOH, 21 h, rt.
Alkylation of 241 with bromoundecanol using the standard conditions proceeded in good yield, as did the subsequent tosylation (scheme 3.19), although poor solubility in DCM necessitated the use of more dilute conditions. In a trial small scale reaction of 243 with potassium thioacetate in PEG 400 overnight no reaction was observed by tlc. Heating for 1 h at 50 - 65 °C did not significantly accelerate the reaction. Addition of methanol and refluxing for 4 h afforded a mixture of thiol, identified by a quartet at 2.51 ppm, and a disulfide (figure 3.14), with a triplet at 2.67 ppm. Another small scale reaction of 243 with potassium thioacetate in methanol, with sufficient THF to solubilize the 243, at room temperature for 2 days afforded a mixture of the desired product 244, disulfide and other products tentatively proposed to be tri- and tetrasulfides (figure 3.14), with triplets at 2.86 and 2.93 ppm respectively. The reaction was repeated on a 3 g scale, with degassing of the THF/MeOH solvent to avoid polysulfide formation, and after one day 244 was isolated in 66 % yield following recrystallization. The byproduct formation observed during trial reactions is likely due to the comparatively low rate of reaction of 243 because of its poor solubility, which allows methanolysis and hydrolysis of 244 to compete with its formation.
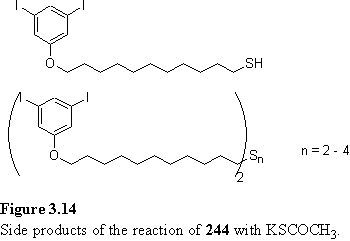
The thioacetate group has been reported to be compatible with Pd catalysed coupling conditions, although the use of the more hindered iPr2NEt instead of Et3N as base was recommended to avoid cleavage of aryl thioacetates.172,396 244 was coupled with acetylene porphyrin 245 to afford 246 after zinc metallation (scheme 3.20). Yields were disappointing, and this can at least in part be attributed to difficulties experienced in purification.
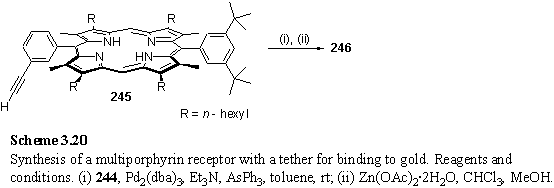
As a model compound with a simpler 1H NMR spectrum, 247 was prepared but again purification was very problematic so as an alternative 249 was synthesized. It was hoped that the benzyl alcohol group would increase the polarity of the product allowing more facile separation from the apolar porphyrin starting material.285 To an extent this was the case, and 248 was isolated satisfactorily pure by NMR in 40 % yield then metallated to 249.
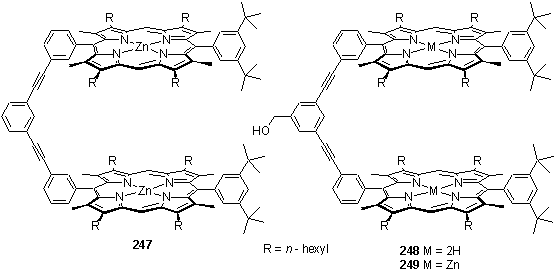
1H NMR titration of 246 with 4,4'-bipyridine in CDCl3 confirmed that the bipyridine is indeed bound by the porphyrins. When less than one equivalent of bipyridine was added the resonances of bound bipyridine were observed at 4.60 (Dd -2.93) and 1.85 (Dd -6.89) ppm. The complexation induced shifts are greater than those experienced by a pyridyl group bound to a single zinc porphyrin397 (-6.0 and -2.2 for the a and b protons respectively). This result would suggest that the bipyridine does bridge between the porphyrins. Addition of further bipyridine caused these peaks to broaden and disappear, due to exchange between 1:1, 1:2 complexes and free ligand. Likewise 4,4'-bipyridine was observed to bind to 249 with the bound ligand resonances appearing at 4.58 and 1.82 ppm. UV titrations should provide quantitative binding constants although these experiments have yet to be carried out.
3.3.11 Porphyrins for orthogonal self assembly
As discussed in chapter 1 orthogonal self assembly has been proposed as a means of orienting large molecules on surfaces patterned with different materials.31-33 Porphyrin oligomers seemed suitable candidates for OSA as they can take the form of a rigid linear rod which may be functionalized at the termini with carboxylic acid and thiol groups such that one end of the molecule binds a gold surface whereas the opposite end is equipped to bind to ITO. Although it would be difficult to experimentally establish that such a molecule did bind in an oriented fashion at the interface between gold and ITO, the molecular orientation, and consequently the elemental depth profile, of a monolayer on gold would be expected to be inverted with respect to the layer formed on ITO. Angular resolved X-ray photoelectron spectroscopy (ARXPS) is a technique that is capable of producing quantitative elemental depth profiles of monolayers.398,399 Photoelectrons emitted by the monolayer are attenuated as they pass through the layer before they escape into the vacuum and are counted by a detector. Electrons emitted from the surface at low angles pass through more of the sample, so are attenuated to a greater extent, making the low angle measurements more sensitive to the elemental composition at the top of the monolayer. The depth profile is calculated from the variation in electron intensity as a function of the take-off angle.
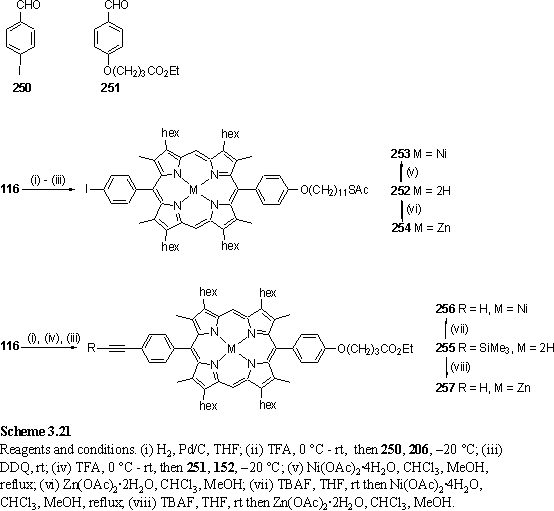
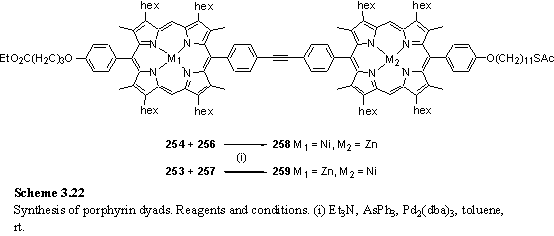
The porphyrin dyads 258 and 259 were designed for OSA on Au and ITO after protecting group cleavage. The unsymmetrical metallation with Zn and Ni was designed with the ARXPS experiment in mind. Both metals give characteristic and easily observed XPS peaks (see section 4.1) and it was hoped that by measurement of these peak intensities as a function of electron take-off angle the orientation of the molecules in a monolayer could be deduced.
The compounds were synthesized according to schemes 3.21 and 3.22 and all steps used routine procedures and intermediates that have been described previously. The alkyl thioacetate was placed on the porphyrin carrying the iodophenyl group as it has been reported that thioacetates may be labile under the conditions required for deprotection of the trimethylsilylacetylene.400 Although initially it was believed that the final Pd coupling had failed due to disappearance of starting materials and appearance of a baseline spot on tlc eluted with hexane/ethyl acetate mixtures it was discovered that the product was not soluble in this eluent which caused it to be retained on the baseline. Elution with toluene, in which the product is soluble, caused the product to move. Column chromatography eluting first with hexane/EA to remove starting materials and AsPh3 followed by toluene/hexane to elute the product proved a convenient means of purifying 258 and 259. Reasonable yields were obtained from the standard coupling conditions with Et3N as a base. The 1H NMR spectrum of 259 is an unremarkable combination of the spectra of the constituent porphyrins, with meso resonances of equal intensity at 10.17 and 9.46 ppm from the Zn and Ni porphyrins respectively. The spectrum of 258 is extremely similar.
Deprotection of 258 and 259 and self assembled monolayer formation have not yet been explored.
The photophysics of 258 and 259 were investigated as part of a collaboration and the results are presented in appendix 2.