3.4 Preparation of Os cluster derivatives
In order to facilitate detection of compounds in self assembled monolayers it is desirable if the compounds possess distinct and intense spectroscopic features which enable them to be distinguished from other organic material which might contaminate a surface. Transition metal carbonyl complexes have been proposed as labelling reagents for biomolecules by virtue of their intense IR absorptions which lie around 2000 cm-1 in a region in which few organic compounds absorb. Subpicomolar quantities of the drug phenobarbital labelled with (h5-C5H4)Mn(CO)5 have been detected in this way401 and analysis of mixtures is possible.402 These compounds also contain a metal atom, which may be detected in trace quantities by techniques sensitive to elemental composition such as XPS and atomic adsorption/emission spectroscopy. This section describes preliminary synthetic efforts to functionalize derivatives of the compounds described in the preceding sections with metal carbonyl clusters to facilitate surface analysis.
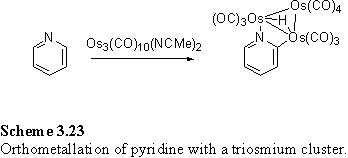
Trinuclear osmium carbonyl, Os3(CO)12, after activation by oxidative decarbonylation with Me3NO in MeCN to form Os3(CO)10(NCMe)2 coordinates to pyridyl groups, followed by orthometallation to furnish a species with a bridging hydride (scheme 3.23).403,404 In collaboration with P. Goh this reaction was attempted with 180 and after preparative tlc 260 was isolated. The 1H NMR spectrum of the compound displays the characteristic bridging hydride singlet at -14.79 ppm. The disulfide group was still intact, despite the possibility of Os coordination, as evidenced by the CH2S triplet at 2.66 ppm, which remained unchanged from its appearance in the starting material 180.
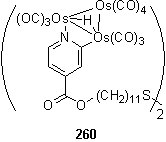
The IR spectrum of 260 is dominated by intense terminal metal carbonyl stretches between 1975 and 2104 cm-1.
The orthometallation of pyridyl porphyrins with trinuclear Os cluster has been demonstrated by Darling.405 With a view to preparation of porphyrins with both thiol and Os cluster functionality, 261 was prepared from 200 and pyridine-4-carbaldehyde using the standard conditions. The thioacetate group was deprotected by aminolysis with pyrrolidine382 to yield thiol 262 in 48 % yield and disulfide 265. The disulfide could be reduced to thiol by heating at 45 °C in a chloroform solution with DTT and Et3N for 1 h.
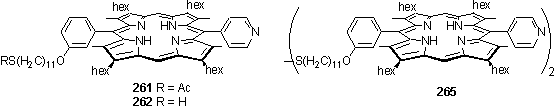
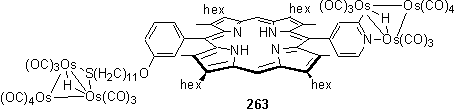
Upon reaction of 262 with Os3(CO)10(NCMe)2 a pair of products was isolated after a troublesome purification by column and plate chromatography. The least polar of the porphyrinic products, 263, displayed two 1H metal hydride resonances in the NMR spectrum at -17.43 and -14.84 ppm and was assigned to a species in which clusters have attacked both the thiol and pyridyl groups. The more upfield hydride resonance is assigned to the S appended cluster. This type of cluster, with a bridging sulfur atom, has been described previously in the literature.406-408 As was observed in the cluster porphyrins of Darling,405 the presence of the cluster on the pyridyl group, which does not lie in the plane of the aromatic ring, desymmetrizes the porphyrin and this is manifested by a splitting of the b-CH3 resonances adjacent to the pyridyl group. In the NMR spectrum of 263 a 3H singlet was observed at 2.78 ppm and this is assigned to the b-CH3 adjacent to the pyridyl group on the same face as the bridging hydride.405 A 5H multiplet at 2.32 ppm is assigned to the b-CH3 on the same face as the Os(CO)4 group, overlapping with the CH2S triplet. The elemental analysis of 263 is fully consistent with the proposed composition.
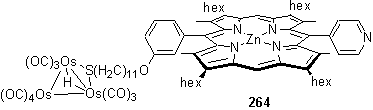
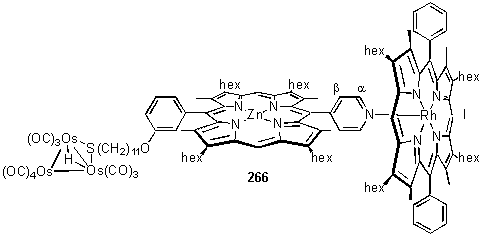
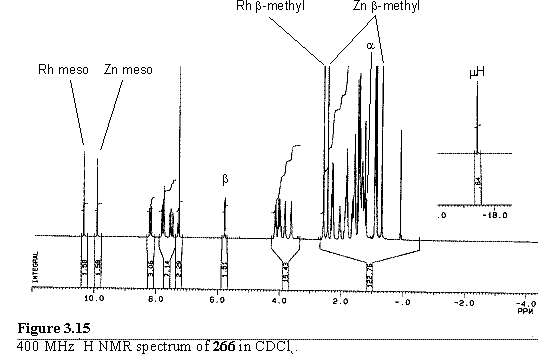
The second product, after zinc metallation, was ascribed to 264 in which the cluster is solely bound to the S atom, with the bridging hydride resonance appearing at -17.44 ppm. In CDCl3 solution the 1H NMR spectrum is broadened due to exchange processes and possibly aggregation due to self association by axial coordination of the pyridyl group to the Zn centre. Addition of Rh porphyrin 118 which binds to the pyridyl group considerably stronger than a Zn porphyrin, caused the predominant structure in solution to be that of 266. The 1H NMR spectrum of 266 (figure 3.15) is sharp and shows the resonances characteristic of axial coordination of a pyridyl porphyrin to 118, as discussed in section 2.5.2.
Evidently the thiol displayed greater reactivity towards the Os cluster than the pyridyl group. The successful isolation of 260 suggests that it may still be possible to isolate products with exclusively orthometallation of the pyridyl group by reaction of disulfide 265 with Os(CO)10(NCMe)2. This reaction was attempted once but led to a mixture of products which were problematic to separate. Side reactions involving coordination of the Os to the pyrrole nitrogens are unlikely to be the cause as Darling found that orthometallation yields were similar for both free-base and Zn pyridyl porphyrins.409
Although 263 and 264 were not the intended products they may be of some interest as clusters are often used as models for surfaces - the so called ‘cluster - surface analogy’.410,411 Thus Adams et al. appended an Os3 cluster to a thiol functionalized ‘molecular wire’ to mimic binding to a metal surface.412