2.7 Rh porphyrin hydrazine coordination chemistry
There have been few literature reports of porphyrin coordination to hydrazines and substituted hydrazines. Collman reported the preparation and redox chemistry of bridged Ru(II) porphyrin dimers303-305 in which hydrazine bound between the already cofacial porphyrins. Hydrazine binding to steroid capped Zn(II) porphyrins has been described,306 and accounts of the syntheses of Ru(IV) hydrazido (-1) and Ti(IV) hydrazido (-2) porphyrins have been published recently.307,308
Continuing the theme of assembly of multiple porphyrin structures using Rh coordination chemistry, it was decided to investigate the interaction of hydrazines with Rh(III) porphyrin.
2.7.1 Rh porphyrin hydrazine complexes
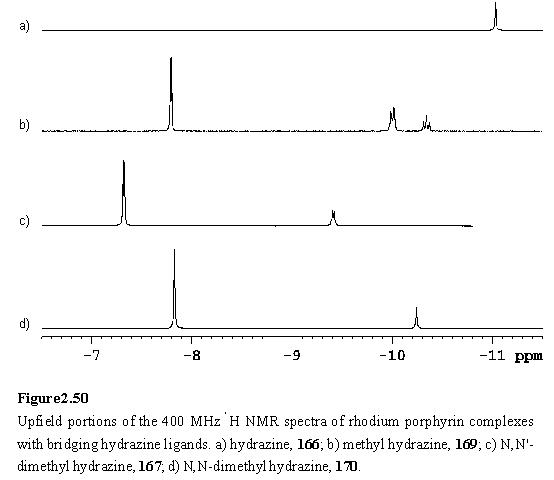
In an initial attempt at preparation of a complex of 118 with hydrazine, a solution of 118 was treated with a large excess of hydrazine monohydrate (11 eq) in DCM, followed by removal of volatiles in vacuo. Although tlc with a range of solvents showed a single spot which was not the starting material, the 1H NMR spectrum indicated the sample to contain a mixture of two species including the bridged complex 166, identified by the highly shielded NH2 resonance at -11.03 ppm. Collman’s cofacial Ru dimer with bridging hydrazine displayed comparably shielded resonances.304,305
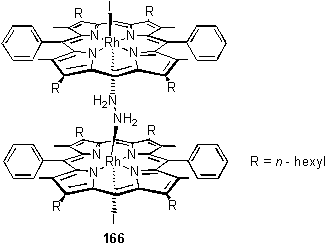
Addition of further portions of 118 to this sample resulted in the disappearance of peaks assigned (vide infra) to the 1:1 complex of 118 with hydrazine, to leave only 166 and a slight excess of 118. 166 appeared to be stable in solution, the NMR spectrum being unchanged after one week at room temperature. Addition of 122 resulted in appearance of many additional resonances in the spectrum which was acquired in less than one hour from mixing. 4 meso peaks could be resolved, those at 10.30 and 10.31 ppm corresponding to free 118 and 122 and those at 9.39 and 9.38 ppm to dimeric complexes with bridging hydrazine. Below 0 ppm the spectrum was clear, except for the NH2 resonances at -10.97 and -10.99 ppm. The presence of three CO2CH3 resonances (4.21 ppm and two closely spaced peaks at 4.13 ppm) indicates that exchange of the porphyrins produced all of the possible complexes containing porphyrin 122. The spectrum was unchanged after 24 hours demonstrating that the exchange is complete within minutes or less. Since the solution always contained a slight excess of porphyrin, a dissociative exchange mechanism appears likely, in which the 1:1 porphyrin:hydrazine complex is an intermediate. The concentration of this postulated intermediate was too low to be detected by NMR.
An 1H NMR titration of 118·MeOH with hydrazine monohydrate in CDCl3 was carried out (figure 2.48). The solubility of the hydrazine in CDCl3 at 0.07 M appeared sufficient, although attempts at making solutions of higher concentration were hampered by emulsion formation. During the titration, addition of < 0.5 eq hydrazine yielded 166 as the only hydrazine containing species. However as more hydrazine was added 166 broke up to a 1:1 complex. Disappointingly it proved difficult to quantify the data and calculate a reliably. The cause of these problems was attributed to uncertainty in the determination of the concentration of unbound hydrazine, which cannot be directly measured by integration.
The peak at -3.28 ppm assigned to the unbound NH2 group of the 1:1 complex broadened with progressive addition of hydrazine, whereas that at -3.77 ppm assigned to the Rh bound NH2 group remained sharp. This is proposed to be due to exchange of the NH2 protons with those of excess hydrazine and water in the solvent, catalysed by traces of acidic impurities.309 The bound NH2 does not have a free lone pair which can become protonated enabling the exchange to occur. Exposure of the sample to TFA vapour further broadened the -3.28 ppm peak, supporting this hypothesis.
The monomer/dimer equilibrium in solution could be shifted towards dimer 166 by passage of the solution through a silica plug eluted with DCM/hexane (1/1), which serves to remove hydrazine. In this way a mixture with initial meso resonances in the ratio 1:0.77 monomer:dimer was converted to a mixture in which the resonances were in the ratio 1:9.6. This also removed several minor unidentified impurities, presumably polar material which was retained on the silica gel. 8.4 mg of material was recovered after this treatment, starting from 10 mg of 118·MeOH, confirming that filtration through silica does not simply remove the 1:1 complex to leave 166.
Similarly, crystallization of a mixture of the 1:1 and 2:1 complexes from toluene and methanol afforded X-ray quality crystals of 166. During data collection with a laboratory X-ray source these crystals ceased to diffract, possibly due to a phase transition. More rapid data collection with synchrotron X-rays was successful and the structure is shown in figure 2.49.
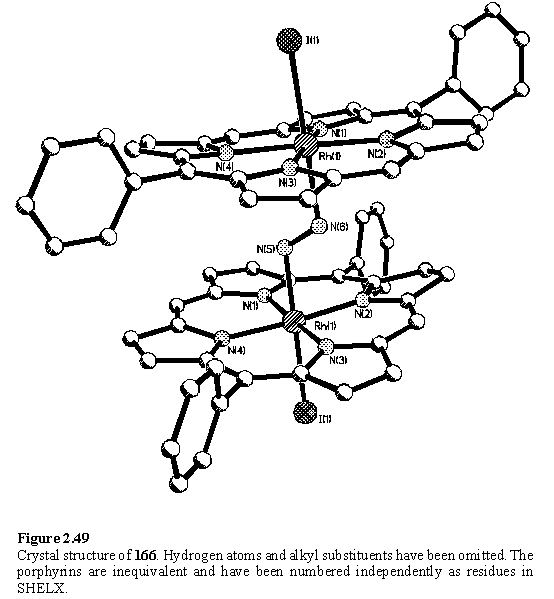
In the solid state the hydrazine adopts a trans geometry with a Rh-N-N-Rh torsion angle of 178.5(1)°. The N-N bond length of 1.47(1) Å is in the range typically reported for bridging hydrazine complexes.310 The porphyrins are oriented with almost parallel planes, but twisted slightly, apparently to avoid steric clash of the peripheral substituents. The porphyrins themselves are distorted from planarity although these distortions are irregular and not easily classified.
A preparative route to 166 was found to be treatment of 118 in DCM with ~1 eq hydrazine in water, followed by a standard aqueous work-up. Evidently the remaining 0.5 eq of hydrazine is removed in the aqueous phase. Material thus prepared was used to obtain an IR spectrum. A weak peak at 3231 cm-1 was assigned311 to the NH2 stretch of the hydrazine.
2.7.2 Rh porphyrin complexes with substituted hydrazines
N,N'-Dimethyl hydrazine is commercially available as the dihydrochloride salt. Extraction of an aqueous solution of this with 118 in DCM afforded mostly the starting material, with traces of dimer 167 evidenced by peaks at -9.40 and -7.32 ppm in the proton NMR spectrum (figure 2.50 c).
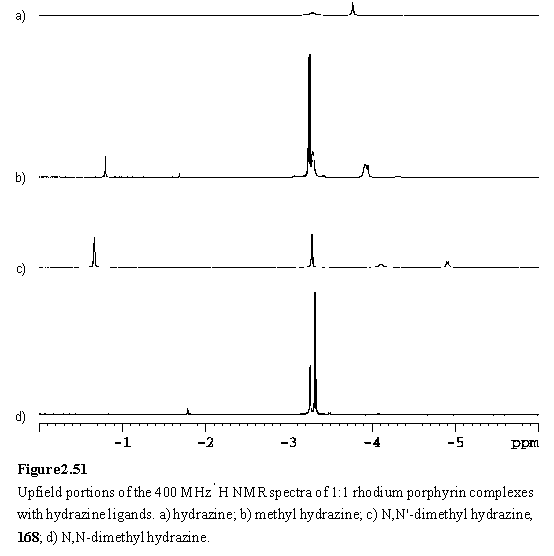
However if the aqueous phase was basified by addition of several drops of 3 N NaOH, the 1:1 complex 168 was obtained. Both the 1H and 13C NMR spectra of 168 were complicated by the formation of a chiral centre at the bound nitrogen of the hydrazine. This removes all mirror symmetry from the porphyrin, leaving only pseudo C2 symmetry (assuming that there is rotation about the Rh-Nax bond), which leads to splittings of the hexyl and b-methyl resonances but not the meso or aryl resonances. Approximate complexation induced shifts (figure 2.54) have been calculated for the hydrazine methyl groups from a spectrum reported for the free ligand in CCl4.312 Coupling is observed between the proton of the coordinated nitrogen and its methyl group with 3JHNCH = 6 Hz (figure 2.51 c). The 3JHNCH coupling constant in free N,N'-dimethyl hydrazine has been reported to be 6.1 Hz.313 The other methyl group of the ligand appears as a singlet presumably because exchange of the proton at the unbound nitrogen occurs too fast to observe any coupling.
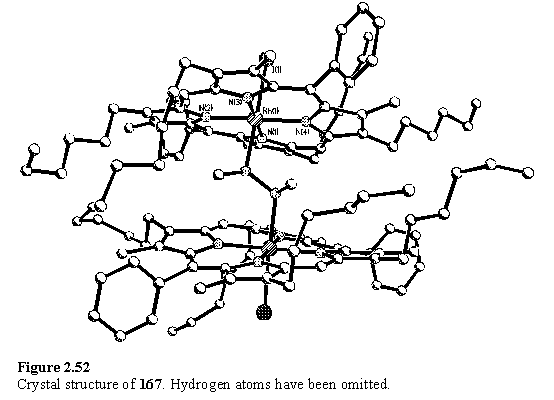
Passing this sample through silica, eluting with DCM/hexane (1/1), followed by recrystallization from chloroform/methanol converted 168 to 167. X-ray quality crystals were obtained by a further recrystallization. Although dimer 167 could exist as a mixture of diastereomers, NMR suggested that only one was present. A single meso, and doubled b-methyl and hexyl resonances were observed, consistent with average C2 symmetry. However fast interconversion of diastereomers on the NMR time-scale cannot be ruled out. The methyl groups of the hydrazine once again appear as a doublet with 3JHNCH = 6 Hz. In the solid state structure (figure 2.52) the ligand is ordered and the chirality of each of the nitrogen atoms in a molecule is the same, but the crystal is centrosymmetric, with a molecule of each enantiomer in the unit cell. The porphyrins are not parallel, but tilted to accommodate the methyl groups of the ligand, and are rotated at ~90° to each other to avoid steric clash of the substituents. The angle between the best fit porphyrin planes is 28.31(5)°.
The hydrazines MeNHNH2 and Me2NNH2 are commercially available liquids and NMR titrations of these were carried out with 118·MeOH in CDCl3. On addition of < 0.5 eq of the hydrazine, the expected bridging complexes, 169 and 170, were observed as evidenced by highly shielded NH and methyl resonances (figure 2.50 b, d). The resonances of MeNHNH2 all appear as multiplets. The Me group is split into a doublet with 3JHNCH = 6 Hz, compared to literature values of 6.3 Hz for free methyl hydrazine,314 and 5.8 Hz for a 1:1 adduct with BMe3.315 The NH2 resonance also appears as a doublet, with a splitting of 12 Hz. This splitting may be due to 3JHNNH or to inequivalence of these diastereotopic protons. 3JHNNH was reported as 6.25 Hz for free methyl hydrazine,314 and 4.5 Hz for the BMe3 complex.315 The NH proton resonance resembles a triplet with a splitting of 12 Hz, instead of a predicted 12 line multiplet. The reason for the lack of appearance of the 6 Hz coupling to the methyl protons is not clear. Additional complications to the spectrum may arise from coupling to the Rh nucleus, and from the quadrupole moment of the nitrogen nucleus. Spectra of substituted hydrazines reported in the literature were acquired with 14N decoupling to eliminate the latter effect.313,314
Further addition of the hydrazines dissociated the dimers to yield 1:1 complexes. However the situation is complicated by the possibility of two isomeric forms in which the hydrazine coordinates through different nitrogen atoms. Theoretical and NMR studies316 have supported the belief that the substituted nitrogen atom is more basic. However in all reported crystal structures317-322 of metal complexes of methyl hydrazine and N,N-dimethyl hydrazine these ligands bind through the unsubstituted and less sterically encumbered nitrogen atom.

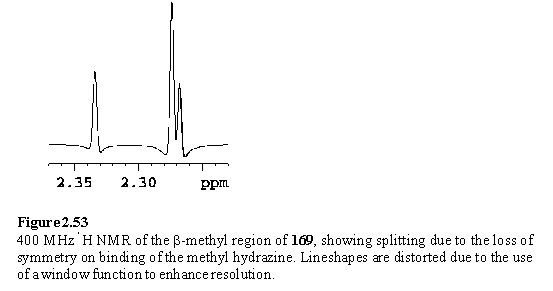
On the basis of the complexation induced shifts (figure 2.54) of the methyl groups, Me2NNH2 appeared to bind to 118 preferentially through the alkylated nitrogen atom. The approach of the methyl groups to the porphyrin does not sterically inhibit complexation to the more basic nitrogen atom. The situation for MeNHNH2 is not so clear cut (figure 2.51 b). The chemical shift of the methyl group is consistent with assignment of the major species as the Rh-NHMeNH2 complex. Several minor peaks have been tentatively ascribed to the isomeric complex, Rh-NH2NHMe. In contrast to 168, no splitting of the b-methyl resonances is observed due to chirality at the bound nitrogen, although splitting is evident in the b-methyl region of the spectrum of 169 (figure 2.53). Passing the samples containing excess hydrazine through silica gel eluting with DCM/hexane (1/1) afforded 169 and 170.
The IR spectrum of 170 showed a sharp but weak peak at 3216 cm-1, whereas 169 gave a group of peaks at 3224 cm-1 which are attributed to NH stretches by analogy with 166.
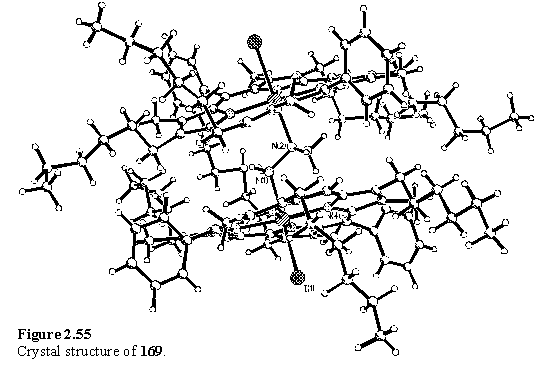
X-ray quality crystals of 169 and 170 were obtained and the structures are shown in figures 2.55 and 2.56. The structure of 169 is extremely similar to that of 167, except the single methyl group is disordered over the pair of sites which are fully occupied in 167. The porphyrins are canted to accommodate the methyl substituents; the angle between the best fit porphyrin planes of 169 is 25.94(3)°. The ligand of 170 is disordered over two sites, with the methyl groups directed at either of the two inequivalent porphyrin units. The structure also resembles that of 167, the porphyrins are canted to the same extent with an angle between the best fit porphyrin planes of 25.49(9)°.
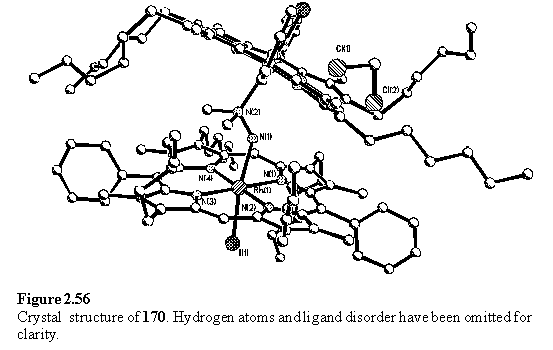
The first attempt at obtaining crystals of 170 by layering a solution containing excess dimethyl hydrazine with methanol produced a mixture of crystals. By diffraction, one of these was identified as the organometallic derivative 123, the structure of which had been previously determined (figure 2.13). Presumably the methyl group originates from decomposition of the methyl hydrazine. The other crystals were not of sufficient quality for structure determination. However NMR of the crystals, redissolved in CDCl3, confirmed the presence of 123 and 170. To investigate the cause of the decomposition 118·THF was treated with 10 eq Me2NNH2 in CDCl3. The sample was divided into four portions in NMR tubes which were exposed to combinations of air and light. Samples not exposed to air were sealed with septa, freeze-thaw degassed and placed under nitrogen. The other sample tubes were closed with ordinary caps. Ambient light was excluded by the use of brown glass tubes, wrapped in foil. After two days an 1H NMR spectrum was acquired of each sample. The sample exposed to both air and light exhibited seven meso peaks and numerous peaks below 0 ppm, implying extensive decomposition. In contrast the other samples appeared to be largely the 1:1 complex of 118 with Me2NNH2. After passing through a silica plug, which retained dark coloured material on the baseline, the sample exposed to air and light consisted largely of 123, by comparison with the spectrum of an authentic sample. The other samples were converted to 170, with the sample from which air and light had been excluded being the cleanest. In the gas and solution phases the oxidation of N,N-dimethyl hydrazine by oxygen has been proposed to occur by a series of radical reactions leading to the formation of dimethyl diazene which undergoes further decomposition to produce a wide variety of products including nitrogen, methane, ammonia and formaldehyde dimethylhydrazone.323 Reaction of these intermediates with 118 is likely to lead to the observed product although the mechanism is uncertain. Similar alkylated Rh products have been observed as products of the reaction of RhCl3·xH2O with free-base porphyrins in hot amide solvents, and were believed to arise by initial porphyrin metallation followed by reaction with the solvent.324, 325