2.8 Rhodium porphyrin coordination to sulfur and selenium ligands
Rh(III) porphyrins are predicted to bind to ‘soft’ donor atoms such as sulfur, selenium and phosphorus. To the best of our knowledge the coordination chemistry of Rh porphyrins with sulfur and selenium ligands had not been explored. This area was investigated, both with a view to assessing the potential of this chemistry for assembling porphyrin nanostructures, and because of the implications for preparation of thiol derivatized Rh porphyrin self assembled monolayers on gold.
NMR and UV titration experiments were performed in which a sulfur or selenium ligand was added to 118·MeOH. NMR experiments were performed in CDCl3. Binding constants cannot be extracted readily from the titration data, because of competition of the added ligand with MeOH and water. In all cases shielded resonances assigned to a bound ligand were observed, and the intensities and complexation induced shifts have enabled the stoichiometry and site of binding to be established.
UV titrations were carried out initially using freshly distilled DCM as solvent. However the Soret band of recrystallized 118 in dry DCM at a concentration of 3 - 5 mM was observed at 402 nm with a shoulder at longer wavelength. On addition of MeOH a single Soret band at 410 nm was observed. In neat DCM the shoulder is likely to correspond to the MeOH or water complex of 118, and the main band to a porphyrin with 5 coordinate Rh. To avoid the presence of multiple porphyrin coordination species, a mixed DCM/MeOH solvent system was chosen, such that in the absence of any additional ligand, the porphyrin would exist largely as the MeOH complex. Binding constants determined in this solvent system correspond to displacement of MeOH by the ligand. The data are comparable provided that the same solvent composition is maintained for all titrations. UV titration data were analysed by the program Specfit.257
2.8.1 Thiols
The complex of 118 with EtSH, 171, was prepared by treatment of a chloroform solution of 118 with EtSH, followed by drying in vacuo. Loss of HI to leave a coordinated thiolate was considered as a possibility, but this was ruled out on the basis of the NMR spectrum in which the SH proton is clearly visible as a 1H triplet (figure 2.57), also confirming the 1:1 stoichiometry of the complex.
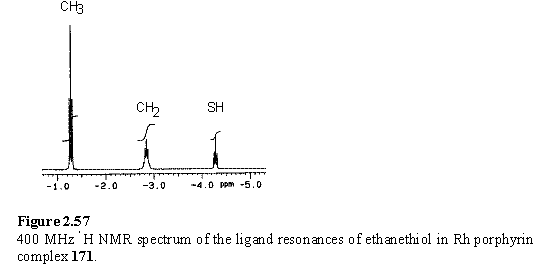
The binding constant (table 2.3) (in competition with MeOH) of 118 with EtSH was determined by UV titration, and an acceptable fit of calculated and experimental data was obtained.
|
Ligand |
log (K) |
|
Me2S |
> 7 |
|
DMSO |
6.7 |
|
EtSH |
5.6 |
|
Me2S2 |
5.7 |
|
Me2Se2 |
6.6 |
Table 2.3
Binding constants of 1:1 complexes of sulfur ligands with 118 in DCM/MeOH at 25 °C, as determined by UV/vis spectroscopy. The data for Me2Se2 were fitted with a model including a 2 porphyrin:1 ligand complex, with log(K) = 5.4 for the second porphyrin binding event.
Attempts at obtaining crystals were unsuccessful. The use of MeOH as a non-solvent resulted in formation of crystals of 118·MeOH by displacement of the original thiol ligand by the large excess of MeOH.
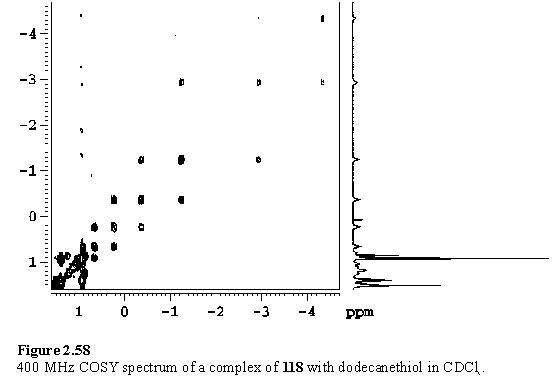
118 was titrated with dodecanethiol in CDCl3. The ability of the porphyrin to act as a shift reagent is evident, with the resonances of the ligand shielded and dispersed (figure 2.58). Broadened resonances resulted on addition of > 1 eq dodecanethiol due to exchange of free and bound ligand.
1H NMR titration of 4·MeOH with benzene thiol revealed formation of the expected 1:1 complex. Sharp ligand resonances were observed when < 1 eq PhSH had been added but these broadened as more ligand was added. A binding constant could not be calculated from the UV titration data, as negligible binding was observed at the concentrations employed. The weak binding, relative to EtSH, is likely the result of the electronic effect of the phenyl group which is conjugated to the lone pairs of the sulfur atom and reduces their donor ability.
2.8.2 Sulfoxides
118·MeOH was titrated with DMSO in CDCl3 to form 172. No change was observed in the meso proton resonance but a bound 6H ligand peak appeared at -2.23 ppm. This peak remained sharp even in the presence of excess DMSO. The large shielding of the methyl groups is consistent with binding of the ligand through the sulfur atom, and not through oxygen. However the presence of an O coordinated species in fast exchange with an S coordinated form cannot be ruled out from this experiment.
The binding constant was estimated by UV titration (table 2.3), although the calculated and experimental data were not in perfect agreement. The reason for this is unclear, but could be due to the existence of species not included in the model or simply experimental error. In support of the former explanation was the appearance of small peaks in the NMR spectrum downfield of the main meso and bound DMSO resonances.
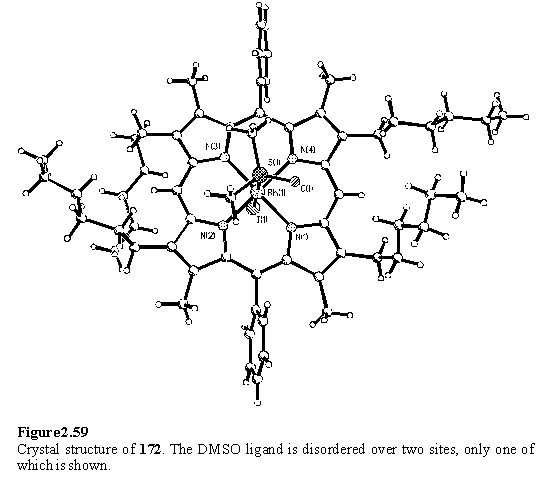
The crystal structure of 172 was obtained (figure 2.59) by crystallization from a DCM solution layered with MeOH and DMSO. As expected on the basis of the NMR spectrum, the DMSO is bound through the sulfur atom with a Rh - S distance of 2.317(1) Å.
Binding of diphenyl sulfoxide was observed by 1H NMR but the ligand peaks broadened and shifted when excess ligand was added. No binding was observed at the concentration employed for UV titration.
2.8.3 Sulfides
A sample of the complex 173 was prepared by reaction of 118 with Me2S followed by filtration though a silica plug, eluted with a DCM/hexane mixture. The ligand was observed by 1H NMR as a sharp singlet at -3.12 ppm. The binding constant can only be estimated by UV titration as > 107 M-1, due to the sharpness of the titration curve.
In a competitive binding experiment, 124 was titrated with Me2S in CDCl3 and monitored by 1H NMR. Pyridine was displaced to a significant extent, and by integration of the resonances of bound Me2S, bound pyridine and free Me2S an equilibrium constant of K = 0.4 was determined for the equilibrium:
![]()
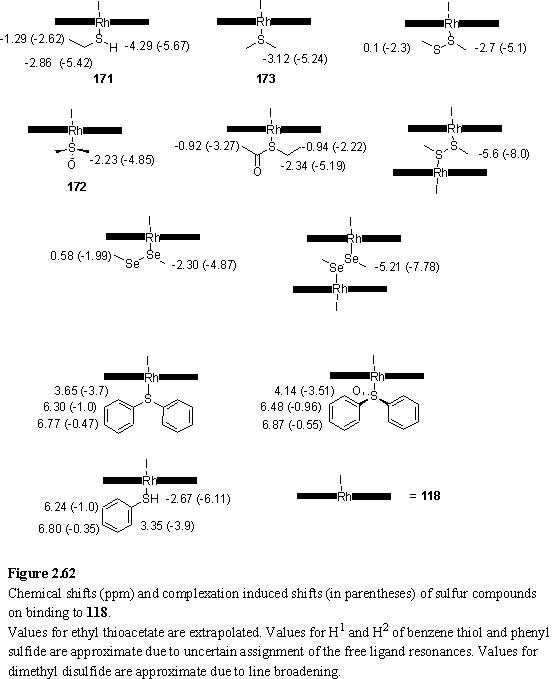
Although this alkyl sulfide was found to bind with a strength comparable to pyridine, the aryl sulfide Ph2S displayed weak binding (by UV), consistent with the trend observed for thiols and sulfoxides. However binding was observed by 1H NMR, and the chemical shifts of the bound ligand are given in figure 2.62.
2.8.4 Disulfides
Disulfides, by analogy with hydrazines, could potentially act as bridging ligands between two porphyrins. A number of crystal structures in which Me2S2 bridges between transition metals326-328 have been described in the literature.
Me2S2 was demonstrated to act as a bridging ligand for 118 by 1H NMR titration. Although peaks were broad, evidence for the bridged dimer came from an upfield shift of the porphyrin meso resonance to 9.8 ppm, and the appearance of a peak at -5.6 ppm from the methyl groups of the ligand. On addition of > 0.5 eq of Me2S2 two new resonances appeared, assigned to the ligand methyl groups of a 1:1 complex (figure 2.62), and the -5.6 ppm peak disappeared. The meso resonance sharpened and shifted downfield to 10.22 ppm as the 2:1 complex was broken up. The complexation induced shifts are non-additive, most likely due to the electronic and conformational changes of the ligand on binding to the porphyrins.
UV titration data were fitted (table 2.3) to a model containing only a 1:1 complex as fitting to a model with both 2:1 and 1:1 species gave implausible results. The concentration of the 2:1 species may be insignificant due to the high dilution of the UV experiment.
2.8.5 Diselenides
Me2Se2 is also known to act as a bridging ligand between metal centres,329,330 so NMR and UV titrations of 118 were carried out with Me2Se2. The bridging complex of Me2Se2 with 118 was observed by NMR. Resonances were sharper than for the sulfur analogue, and separate meso peaks were observed for each porphyrin species (118, 118·Me2Se2 and 1182·Me2Se2), indicating a lower exchange rate of the selenide ligand. The complexation induced shifts (figure 2.62) of Me2Se2 are slightly smaller than those of Me2S2 reflecting the larger size of the Se atom which places the methyl groups further from the porphyrin plane.
UV titration data gave a poor fit to a 1:1 model. Inclusion of the bridging 2:1 complex in the model improved the fit, while still maintaining realistic calculated spectra and concentration profiles (figures 2.60, 2.61). The binding constants (table 2.3) indicate binding to the selenium atoms is weakly negatively cooperative. The 1:1 binding constant of the diselenide is an order of magnitude higher than that of the disulfide. Although the ‘soft’ rhodium centre is expected to have a greater affinity for the ‘softer’ selenium donor, the lower binding constant may reflect differences in the solvation of the free ligands and not the strength of the Rh-S versus Rh-Se bonds.
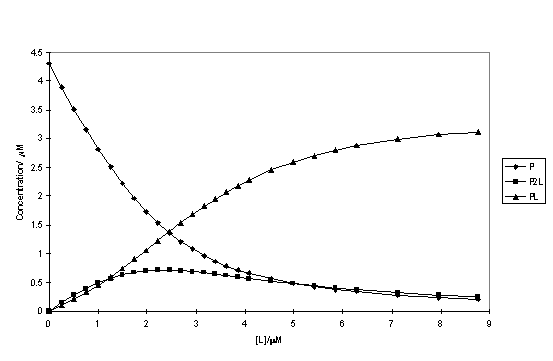
Figure 2.60
Calculated concentrations of complexes of 118 (P) with Me2Se2 (L) in DCM/MeOH as a function of the total ligand concentration.
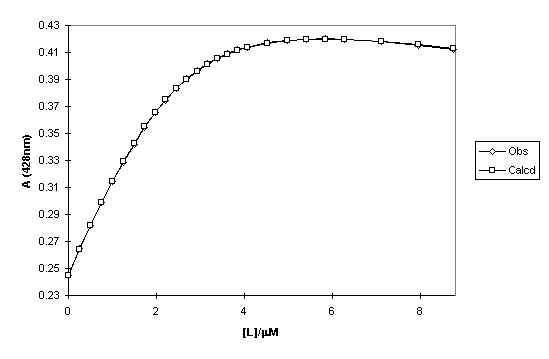
Figure 2.61
Observed and calculated absorbance at 428 nm of a titration of 118 with Me2Se2 (L) in DCM/MeOH as a function of the total ligand concentration.
2.8.6 Thioesters
Binding of ethylthioacetate to 118 was demonstrated by 1H NMR titration. Broadened ligand peaks which shifted downfield during the titration were observed indicating fast exchange of free and bound ligand. Complexation induced shifts (figure 2.62) have been estimated by extrapolation. The large shift of the CH2S resonance indicates that binding occurs through the sulfur atom, although the existence of an oxygen coordinated species cannot be ruled out. The shift of the CH3CO resonance also seems large suggesting the coordination geometry causes this methyl group to approach the porphyrin plane. Further evidence for coordination of thioacetate to Rh(III) porphyrin through the sulfur atom is presented in section 3.3.3.
2.8.7 Summary
In conclusion, Rh(III) porphyrin was found to bind to a wide variety of sulfur and selenium compounds with a range of binding constants. The association constants for 1:1 complexes were found to vary in the order Me2S > DMSO » Me2Se2 > EtSH » Me2S2. The disulfides and diselenides were also found to act as bridging ligands between two porphyrins. Aromatic substitution at sulfur resulted in weaker binding. Sulfoxides and thioesters appeared to bind through the sulfur atom, and not through oxygen.
The lability of many of the ligands was evident from line broadening and fast exchange in the 400 MHz 1H NMR spectra at room temperature.
Of the ligands studied, none with the possible exception of the alkyl sulfide and sulfoxide, seemed to possess such favourable properties as pyridines for assembly of supramolecular species such as the dendrimers described in section 2.6. For this application strong binding and sharp NMR spectra at room temperature are desirable.
However for templated synthesis applications the lower binding constants of the sulfur based ligands may be advantageous, and permit removal of the template after the reaction. Sulfur templates may suffer the disadvantage of being less chemically robust than pyridyl compounds, placing restrictions on the type of reaction that can be templated.