1.5 Peripheral coordination arrays of porphyrins
The porphyrin periphery may be functionalized with metal binding or hydrogen bonding groups that permit assembly into a variety of topologies ranging from discrete linear and cyclic species, to dendrimers, and coordination polymers in one, two and three dimensions.
1.5.1 Discrete linear arrays
Non-cyclic porphyrin arrays may be obtained by appending a porphyrin with a single metal binding or hydrogen bonding motif, so that two such moieties can be linked by a single metal ion, or by interaction of complementary hydrogen bonding groups.
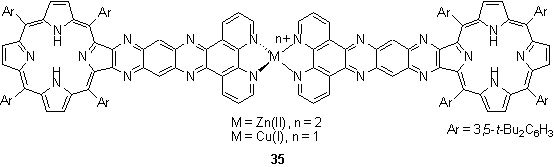
Crossley et al. prepared porphyrins fused to phenanthroline units with a view to electronically coupling the porphyrin p system to an external redox active metal centre.46 Two phenanthroline units coordinated to Zn(II) or Cu(I) ions to produce the dimeric porphyrin arrays 35. The arrays were prepared by addition of 0.5 eq of Zn(OAc)2 or [Cu(NCMe)4]PF6 to the monomeric free-base porphyrin components. Porphyrin metallation was sufficiently slow that it did not compete with coordination of the metal ions to the phenanthroline groups. A similar complex was subsequently reported with a shorter aromatic linker between the porphyrin and phenanthroline units with the aim of both improving solubility and increasing electronic coupling.47
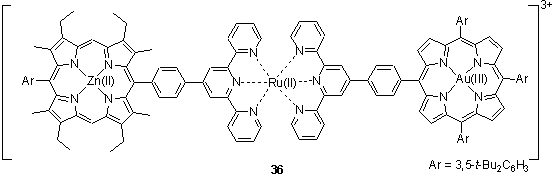
Sauvage and coworkers have described the syntheses of porphyrin dimers linked by a Ru(II) terpyridine complex.48 The terpyridine groups were coordinated in a stepwise manner to the Ru centre which enabled the preparation of unsymmetrical heterometallic dimers such as 36. The singlet excited state of the Zn porphyrin of 36 was found to transfer an electron to the Ru(II) unit. A second electron transfer to the Au(III) porphyrin competed with back transfer to the Zn porphyrin radical cation.49
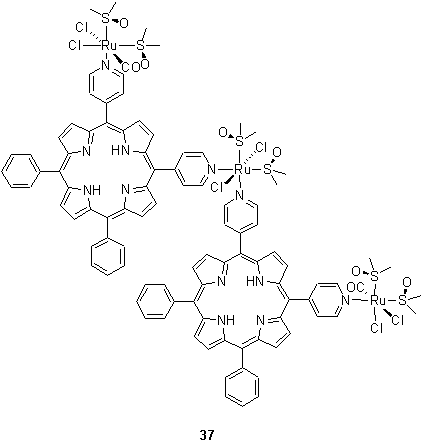
Alessio et al. have used the coordination of pyridyl porphyrins to a variety of Ru(II) DMSO and carbonyl complexes to prepare porphyrin dimers, for example 37, in which the porphyrins adopt a cis geometry at the Ru centre.50,51 Similar structures in which two or four pyridyl porphyrins have been assembled using square planar Pd(II) and Pt(II) as the metal unit have been reported.52,53
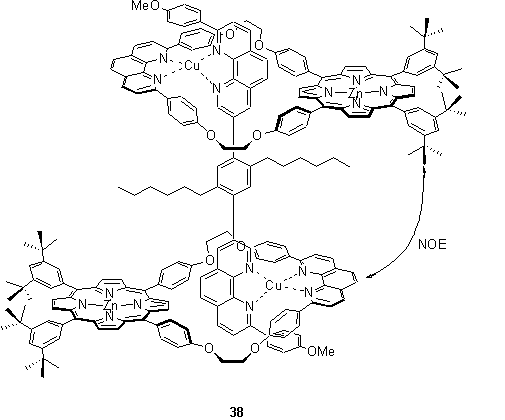
To construct a porphyrin dimer using peripheral metal coordination it is not necessary that both of the porphyrins’ peripheral donor groups coordinate to the same metal ion. Indeed Sauvage, Chambron and coworkers assembled a dimer by threading porphyrin phenanthroline macrocycles around a rod-like bis-chelating ligand with coordination to Cu(I) providing the driving force.54,55 ROESY experiments revealed NOE crosspeaks which indicated that the Zn metallated derivative, 38, adopts a conformation in which the porphyrins are tilted towards, and stack against the adjacent Cu(I) phenanthroline unit. However the NOE crosspeaks were absent in the Au(III) analogue possibly because this stacking is likely to be disfavoured due to electrostatic repulsion between the cationic Au(III) porphyrins and Cu(I) phenanthroline complex.
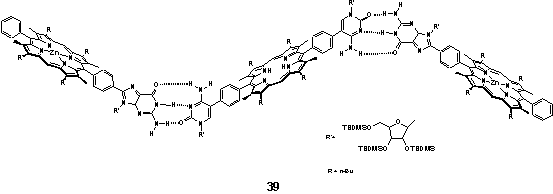
There are comparatively few examples of discrete hydrogen bonded linear arrays of porphyrins. Sessler et al. exploited Watson-Crick nucleobase pairing to assembly a trimeric array 39 in CD2Cl2 as a photosynthetic model in which energy transfer occurs between the zinc and free-base porphyrins.56 The protected sugar unit acts as a solubilizing group.
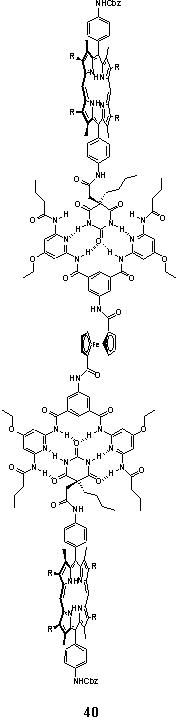
Hydrogen bond mediated assembly of a porphyrin barbituric acid derivative and a pair of 2,6-diaminopyridine units linked to a central redox active ferrocene unit yielded the porphyrin dimer 40.57
1.5.2 Cyclic peripheral coordination assemblies
If a porphyrin carries more than one peripheral ligand or hydrogen bonding site, then formation of cyclic oligomers is a possibility. As bis(4-pyridyl)porphyrins present donor groups 90 or 180° apart these compounds lend themselves to the creation of molecular squares in which porphyrins occupy either vertices or edges respectively.
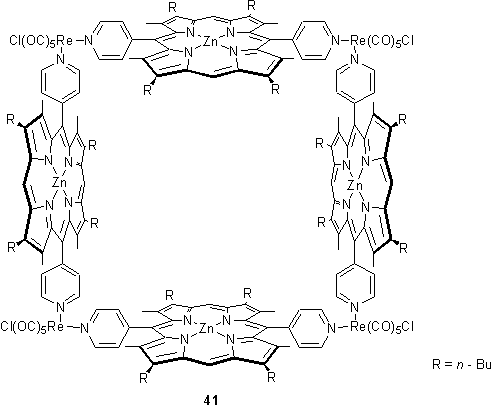
Hupp and coworkers prepared the square 41 in high yield by refluxing the porphyrin component with Re(CO)5Cl in THF/toluene, followed by treatment with zinc acetate to metallate the porphyrins.58 The square was believed to exist in a box-like conformation in which the opposite pairs of porphyrins are cofacial. This hypothesis was supported by a fluorescence titration with 5,10,15,20-tetra(4-pyridyl)porphyrin which quenched the fluorescence of 41 after addition of one equivalent. It was proposed that the tetrapyridylporphyrin binds within the cavity of 41 by axial ligation to the four Zn centres, and the binding constant was calculated to be 4 ´ 107 M-1. The ability of 41 to act as a molecular sieve was tested by evaporative casting of a thin film onto an electrode surface.59 By cyclic voltammetry the film was found to be permeable to a variety of small and medium sized molecules and ions such as I-, Ru(NH3)5(4-picoline)2+ and Co(bipyridine)32+. However large transition metal complexes were blocked, and the cut-off size was approximately equal to the diameter of the cavity of 41 implying that transport through the film might occur via this cavity. Films cast from the complex of 41 with tetrapyridylporphyrin were permeable to I- but not Ru(NH3)5(4-picoline)2+ consistent with the notion that the tetrapyridylporphyrin blocks the pores in the film preventing the passage of larger species to the electrode surface.
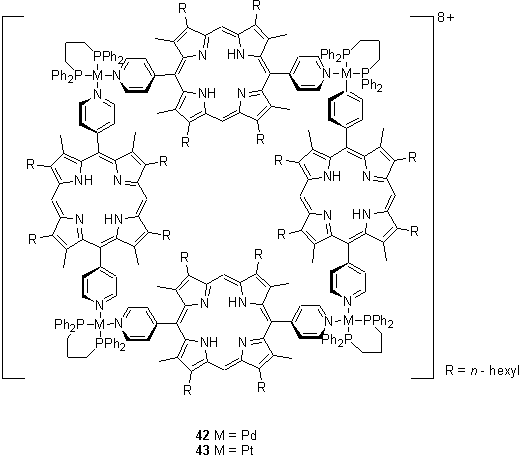
Square planar Pd(II) or Pt(II) with a cis chelating ligand, such as a diphosphine, provides a pair of pyridyl binding sites 90° apart, and thus can act as a vertex of a molecular square. This approach was pursued by Stang et al. who have reported preparation of squares 42 and 43 by treatment of a 5,15-dipyridylporphyrin with a Pd(II) or Pt(II) cis bistriflate.60 The 1H NMR spectrum of these compounds showed two sets of resonances arising from the porphyrin, but one set from the pyridyl groups. This result can be explained by the porphyrins and metal centres adopting a coplanar conformation, with the pyridyl groups perpendicular to this plane, and slow rotation about the M-N and porphyrin-pyridine bonds on the NMR time-scale. The interconversion of the inner and outer porphyrin protons was found to be faster for the Pd containing macrocycles than the Pt analogues.61 If a chiral BINAP ligand was used on the Pd centre then the observation of two peaks in the 31P NMR spectrum combined with the results of molecular modelling studies suggested that the chirality of the BINAP was transferred into puckering of the squares in a D2 symmetric conformation.61 Strong circular dichroism was observed at the wavelength of the porphyrin Soret band.
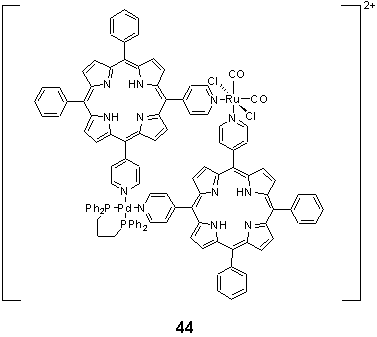
Stepwise coordination of Ru(II) followed by Pd(II) to 5,10-dipyridyl porphyrin furnished a heterobimetallic square, 44. The assembly of the square was monitored by observation of the 1H NMR resonances of the pyridyl protons which were shifted downfield on coordination to the Ru and Pd centres, and the molecular structure corroborated by crystallographic analysis.62
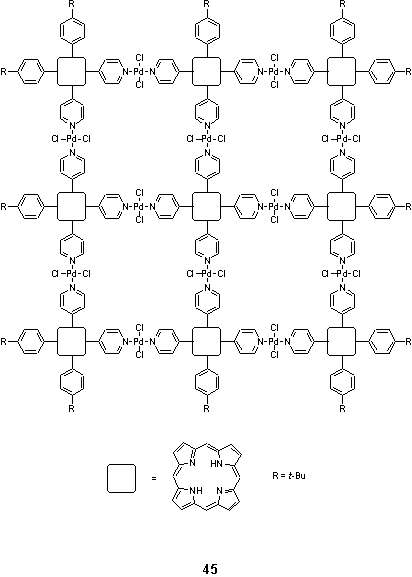
Building on earlier work concerning the self assembly of Pd and Pt linked pyridylporphyrin dimers and tetramers,53 Drain et al. claimed the assembly of a nonameric square array, 45.63 This was prepared by mixing three different pyridyl porphyrin building blocks in the correct stoichiometric ratio with [PdCl2(NCPh)2] in chloroform, with a total porphyrin concentration of < 10 mM to disfavour higher oligomer formation.
Cyclic porphyrin coordination oligomers need not be limited to a square geometry. Substitution of the meta positions of a diaryl porphyrin with acetylene groups that coordinate to trans Pt(II) complexes enabled preparation of an organometallic trimer in which the planes of the three porphyrins are 120° apart.64
1.5.3 Hydrogen bonded cyclic arrays
Drain reported cyclic hydrogen bonded porphyrin arrays including a four component dimer65 and a six component hexamer rosette66 which was assembled from three 5,5-di(butyl)barbituric acid molecules and three bis-porphyrin substituted triaminotriazines.
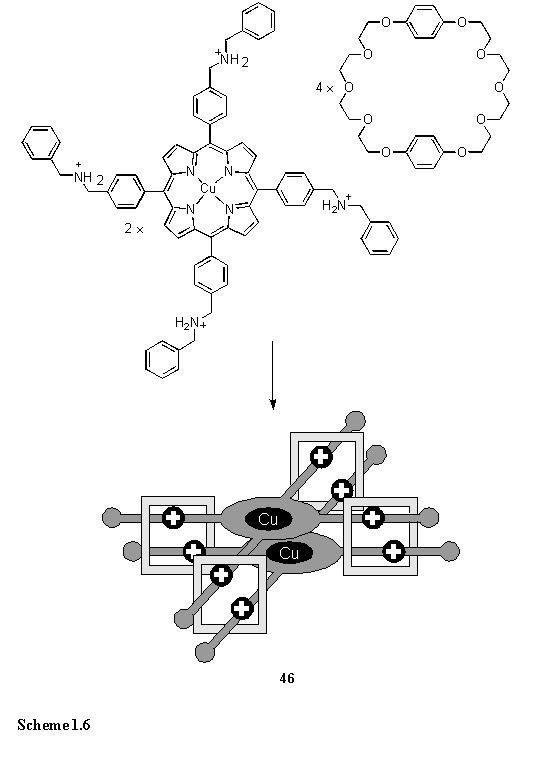
A more unusual porphyrin dimer, 46, assembled by the threading of dibenzylammonium cations through crown ethers and stabilized by NH - O and CH - O hydrogen bonds, was prepared as a mimic of the photosynthetic special pair.67 The ESR spectrum of the Cu(II) porphyrin component in the absence of crown ethers could be interpreted in terms of isolated Cu(II) centres. However on adding the crown ether a change in the ESR spectrum was observed which could be rationalized in terms of a triplet spin system arising from coupling of two Cu(II) ions in close proximity. This observation demonstrated the assembly of the dimer in solution, and the solid state structure was confirmed crystallographically.
1.5.4 Porphyrin peripheral coordination dendrimers
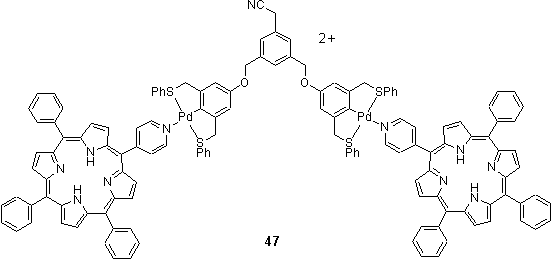
The Pd pincer dendrimer synthesis discussed in section 1.2 was applied to prepare a porphyrin terminated dendrimer.68 After activation of wedge 23 with AgBF4, reaction with two equivalents of pyridyl porphyrin afforded a porphyrin terminated wedge 47 which was coordinated to the core 24 after activation. This sequence of construction was found to be crucial for success, because addition of the pyridyl porphyrin to an activated dendrimer caused scrambling due to displacement of cyano groups from the Pd centres by the more strongly coordinating pyridyl ligand. The authors also reported the preparation of a porphyrin core carrying four Pd pincer complexes around which a porphyrin terminated dendrimer could be assembled.
1.5.5 Peripheral coordination polymers
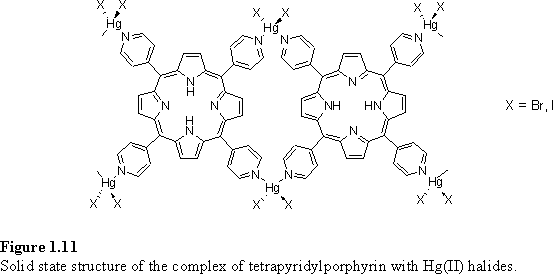
Two recent papers have described the structure (figure 1.11) of a one dimensional coordination polymer formed by linking tetrapyridylporphyrin with approximately tetrahedral HgX2 (X = Br, I) units.69,70 It was found possible to partially Zn metallate the porphyrins without disrupting the structure, either by addition of Zn(NO3)2·6H2O to the solvent used for crystallization, or by directly crystallizing a mixture of free-base and Zn tetrapyridylporphyrin with HgI2. The latter method has the advantage of control over the extent of metallation, and was also found to be applicable to the preparation of crystals containing predetermined percentages of Ni(II) or Cu(II) porphyrin. However if 100 % Zn tetrapyridylporphyrin was used the structure was not preserved due to interference from Zn-pyridine coordination. Another feature of these structures was their ability to include from two to six 1,1,2,2-tetrachloroethane (TCE) solvent molecules per porphyrin by expansion of the distance between the porphyrin layers.
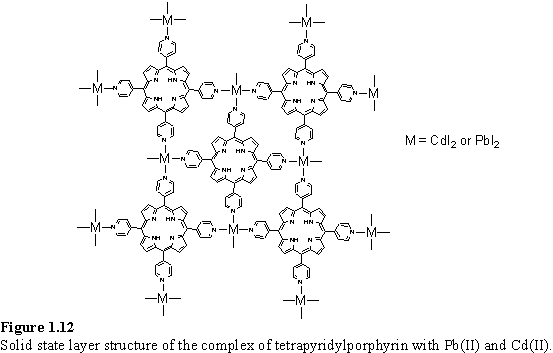
If the tetrapyridylporphyrin was crystallized with eight coordinate Pb(II) or Cd(II) instead of Hg(II) then a two dimensional coordination polymer (figure 1.12) was obtained.70 The halide ions coordinate to the metal ions in a trans manner, perpendicular to the plane of the layer porphyrin with TCE solvent molecules included between the layers.
A three dimensional coordination polymer was produced by evaporation of a solution of Pd-tetrapyridylporphyrin and Cd(NO3)2·4H2O in methanol and water.71 Two pyridyl groups bound to each of two approximately octahedral and inequivalent Cd(II) ions. The pyridyl groups adopted a cis geometry at one Cd centre, but a trans geometry at the other. The result was a structure consisting of linear porphyrin strips connected at opposing vertices by the trans Cd centres. The cis Cd centres link each strip to adjacent crystallographically equivalent strips passing above and below the first strip.
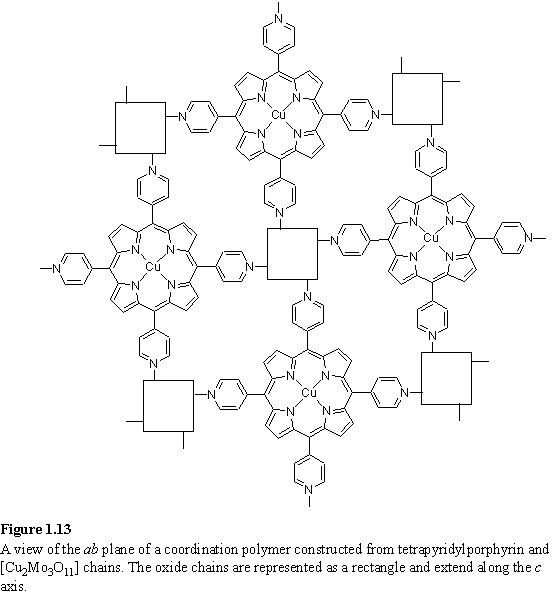
A pair of unusual porphyrin coordination polymers was prepared by hydrothermal reaction of free-base tetrapyridylporphyrin with MoO3 and either Cu(NO3)2·2.5H2O or FeCl2·4H2O.72 The Cu containing product consisted of parallel [Cu2Mo3O11] chains running along the c axis connected by a two dimensional array of Cu(II)-porphyrins in the ab plane (figure 1.13) in which the pyridyl groups coordinate to the Cu(II) ions of the oxide chains. The Fe containing product consisted of [Mo6O19]2- ions surrounded by a network of Fe(II)-porphyrins linked both by axial coordination, and coordination of six pyridyl groups to an Fe(II) ion.
A brief mention must be made of the many crystal structures of porphyrins with peripheral hydrogen bonding groups such as carboxylic acids,73,74 phenols,75,76,77 amides,78 and nitriles.79 In each case hydrogen bonding in the solid state is extensive although rational prediction of the crystal packing from a knowledge of the molecular structure is a non-trivial task due to the complex kinetics and thermodynamics of the crystallization process.