4.1 X-Ray photoelectron spectroscopy (XPS)
The work described in this section was carried out with much assistance from Mr. N. Malone (formerly ICI, Runcorn Heath). Unfortunately this collaboration was terminated prematurely by the break-up of the ICI company, thus leaving some questions unanswered. Additional spectra were measured by Dr. H.-L. Zhang (University of Leeds) at the RUSTI facility at Daresbury laboratory.
XPS was used to characterize a selection of monolayers on gold of the compounds which were available at that time. The spectra were referenced to Au 4f7/2 at a binding energy (BE) of 83.98 eV, as is common in the literature.370
Spectra of monolayers of samples of 180 and 181 which had been synthesized by the triphenylsilanethiol route described in section 3.1.5 were acquired. These spectra displayed the expected peaks arising from ionization of core electrons of C, N, O and S (appendix 3). The resolution of the instrument was insufficient to resolve separate S 2p3/2 and 2p1/2 signals in the noisy data and the S 2p ionization was treated as a single peak centred at BE 163 eV, in reasonable agreement with the value reported for sulfur bound to gold as a thiolate.413,414,422 The C 1s peak appeared at 285 eV, a typical value for aliphatic carbon. Unfortunately the layers also displayed several peaks attributed to contaminants. A peak at 932 eV and a weaker peak at ~20 eV higher energy were assigned to Cu 2p3/2 and Cu 2p1/2 respectively.415 These peaks were also observed on a control sample prepared by dipping a gold substrate into the ethanol that had been used as a solvent for the disulfides. The peaks were not observed on samples for which THF had been used as a solvent or on a ‘clean’ gold sample which had not been exposed to any solvent. Therefore it can be concluded that the ethanol is responsible for the contamination, although it is not clear whether the ethanol contained copper when purchased, or subsequently became impure during handling, despite rigorous cleaning of glassware.
A pair of peaks at 619 eV and ~630 eV was assigned to the I 3d doublet.415 The higher binding energy peak was less intense, consistent with this assignment. These peaks were only observed in the spectra of layers of 180 and 181, and not in any of the control samples indicating the compound samples to be the most likely source of the iodine. As the last step of the preparation of these batches of 180 and 181 was an iodine oxidation it seems plausible that traces of iodine remained in the samples, even though these were analytically pure. Iodide and iodine are known to form chemisorbed adlayers on the Au(111) surface.350
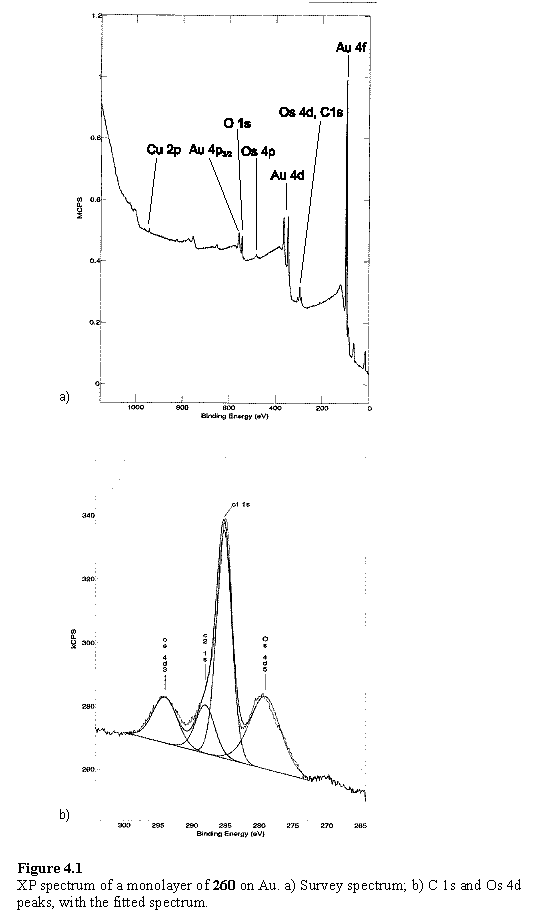
The layer of the triosmium cluster derivative 260 displayed a markedly different spectrum from that of the parent compound 180. Iodine peaks were absent from this sample, but Cu was observed. The Os 4d3/2 and Os 4d5/2 peaks partially overlap with the C 1s peak which made fitting of the spectrum necessary to obtain peak positions and intensities (figure 4.1). The C 1s peak is noticeably asymmetric and was treated as a combination of two peaks. The fitting procedure assigned these energies of 285 and 288 eV, and the Os 4d3/2 and Os 4d5/2 energies of 294 and 279 eV respectively. This is in good agreement with the spectra described in the literature of triosmium clusters immobilized on a SAM416 and for bulk triosmium cluster compunds.417 The C 1s 288 eV component, although at the energy reported for osmium carbonyl carbon, is less intense than the 285 eV component. This is unexpected as there are a similar number of carbonyl and aliphatic carbons in the molecule, and in a monolayer in which the sulfur is bound to gold, the photoelectrons from the alkyl chain would be attenuated by the overlying metal carbonyl component. A possible reason for this apparent anomaly could be a layer of aliphatic organic contaminants adsorbed from the air, over the layer of 260. Organic material was observed on a ‘clean’ gold sample, in the form of a C 1s peak at 285 eV, although relative to the Au 4f this was less intense than observed for samples on which monolayers had intentionally been prepared. Organics are known to adsorb to gold surfaces as soon as they are exposed to the atmosphere, with the level of contamination increasing with increased exposure time.418
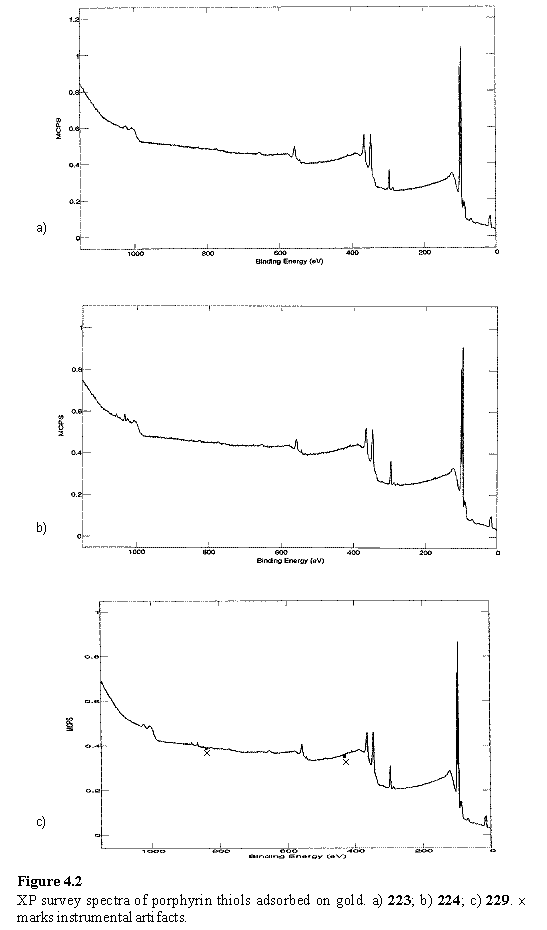
Porphyrin compounds 223, 224, 227, 228 and 229 were adsorbed onto Au from THF solution and examined by XPS. Representative survey spectra are given in figure 4.2. In all cases the Au 4f (doublet at 84 eV) and Au 4p3/2 (547 eV) peaks were clearly visible with similar intensity with respect to the C 1s signal, indicating organic layers of similar thickness as the Au photoelectrons are attenuated during their passage through this layer.419 A weak unresolved S 2p peak was observed in each case at 162 - 163 eV. The layers of 224 and 228 both display peaks at 1022 eV assigned to Zn 2p3/2 in good agreement with other reports of Zn porphyrin monolayers on gold,177 amino terminated glass196 and the bulk.420 The Ni 2p3/2 peak was observed at 855 eV in the layer of 229, in good agreement with the literature.415,420 Neither metal peak was observed in the spectra of the free-base porphyrins 223 and 227 confirming that the origin of the metal is the metalloporphyrin. There is no clear trend in the relative peak intensities (appendix 3), which have been corrected for the ionization cross section of the orbitals involved, although the data were noisy due to time constraints limiting the number of scans that could be acquired. However the relative intensities are approximately in agreement with the elemental compositions of the porphyrins, although it would be unwise to comment in more detail, given the experimental uncertainties and the difficulties inherent in relating peak intensities quantitatively to atomic percentages.419
An attempt at metallation of a monolayer of 227 by immersion of a sample into Zn(OAc)2·2H2O in THF at room temperature for 2 h did not appear to have been successful on the grounds that the Zn 2p peaks were absent from the XP spectrum. This could be simply by virtue of poor solubility of the zinc salt in THF, a slow metallation reaction in this solvent, or as a result of the packing of the porphyrin in the monolayer which renders it inaccessible to metallation, as has been described for porphyrins of a similar structure.174
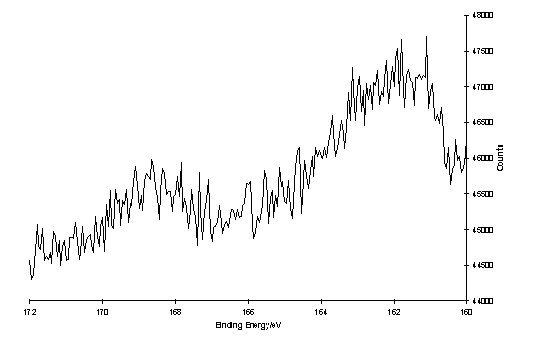
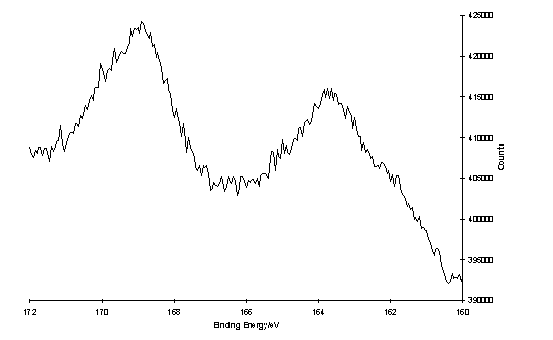
Further XPS measurements of 235 and 224 on gold were made by Dr. H.-L Zhang using a monochromatic X-ray source. The spectrum of 224 acquired with an electron take-off angle of 45° shows an S 2p peak at ~162 eV and an additional peak at ~169 eV (figure 4.3). On decreasing the take-off angle to 10° the intensity of the higher binding energy peak increased relative to the peak at 162 eV, which also appeared to possess a component at ~164 eV which is relatively more intense at this take-off angle. Although curve fitting of these spectra has not been performed a model which accounts for the features observed is a layer of chemisorbed thiolate with an S 2p3/2 binding energy 162 eV, unbound thiol or disulfide at 164 eV, and sulfur species in higher oxidation states at ~169 eV, with the latter two species being located in the upper regions of the surface layer. This assignment is consistent with literature reports of XPS of layers of disulfide containing polymers,421 physisorbed thiols,422 and SAMS oxidized in air423,424 or by direct ozone treatment.425 The layer of 235 showed a single S 2p peak at 168 eV indicating complete oxidation of the sulfur to oxygen containing species (sulfonate, sulfinate). The difference in appearance of the spectra of the adlayers of 224 and 235 indicated that under the conditions which were used for preparation of these layers, identical surface structures were not obtained.
a)
b)
Figure 4.3
S 2p region of the XP spectrum of a layer of oxidized 224 on Au, of a) 45° take-off angle, 1 scan; b) 10° take-off angle, 150 scans.
Rates of oxidation of SAMS are reported to be variable and dependent on surface morphology and monolayer order,423,424 but oxidation can be detectable within hours of exposure to the atmosphere. These spectra were acquired after several days of exposure to air. Ideally samples would be examined immediately after removal from the thiol/disulfide solution, or stored briefly under an inert atmosphere. However due to restricted access to XPS facilities this has not been possible.