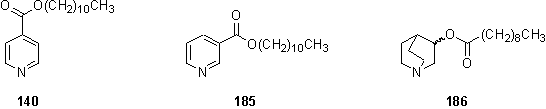3. Synthesis of compounds for self assembly on gold
3.1 Synthesis of ligands
In order to allow the porphyrin self assembly chemistry described in chapter 2 to be carried out on surfaces, with the possibility of two dimensional spatial patterning of the resulting structures, ligands are required with a functional group that binds to the desired surface. The self assembly of thiols or disulfides on gold was chosen as this offers several advantages. Clean gold substrates can be readily prepared by thermal evaporation of gold on to mica or silicon, thus ensuring that surfaces are comparatively free of contamination before adsorption of the sulfur compound. Monolayer preparation does not require use of anhydrous conditions, and the compounds are moderately stable to air, although thiols are prone to oxidation by atmospheric oxygen, especially under basic conditions. Thiol monolayers on the Au(111) surface have been widely studied and a wealth of information is available in the literature.
Alkane thiols with chain lengths greater than around ten carbon atoms are reported to form ordered layers on Au(111).19 It was decided to synthesize compounds with a terminal group capable of coordinating a metalloporphyrin, an alkyl spacer of 11 methylene units, and a thiol or disulfide head group. The choice of the 11 methylene spacer was made on the grounds that this is around the minimum chain length reportedly needed for good ordering, and suitable synthetic precursors, in the form of difunctionalized alkanes, are readily available at modest cost. Both for synthetic convenience and as a spectroscopic handle, a functional group was desired to link the alkyl chain and ligand tail group. Amides were ruled out on the grounds that they can act as hydrogen bond donors and acceptors. This is known to complicate the structure of SAMs,331-335 and could act as another means by which molecules with appropriate functionality could bind to the SAM. Ester or ether groups appeared suitable, combining ease of synthesis and reasonable hydrolytic stability. The carbonyl group of the ester has the additional advantage of a strong IR chromophore at a characteristic frequency, which should assist in analysis of monolayers by reflection - adsorption IR spectroscopy (RAIRS).
3.1.1 Pyridyl ligands
Isonicotinic and nicotinic acid are commercially available building blocks for the preparation of functionalized pyridines. It was decided to derivatize both compounds with alkyl thiols or disulfides, as it was uncertain what orientation the pyridyl group would adopt in self assembled monolayers. The orientation of the head group could be such that the nitrogen atom is unavailable for coordination to a porphyrin.
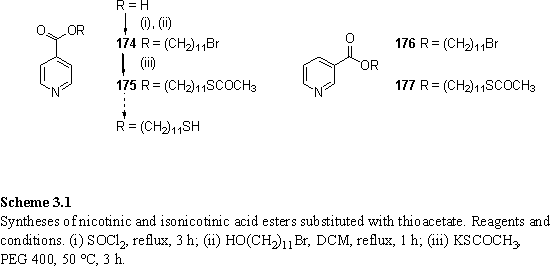
The initial synthetic approach to these compounds (scheme 3.1) was esterification of isonicotinic and nicotinic acids with bromoundecanol, followed by conversion of the bromo group to thiol. The esterification could be performed by refluxing the acid with thionyl chloride, to form an acid chloride, then reaction with 11-bromoundecanol in refluxing DCM. Reaction of the alcohol with commercially available nicotinoyl chloride hydrochloride in DCM and triethylamine also afforded 176, although the purity was found to be inferior to that produced using freshly prepared acid chloride. 174 and 176 were purified from unreacted alcohol by a modified literature procedure336 in which they were precipitated from Et2O as a salt by addition of concentrated sulfuric acid. After deprotonation, traces of yellow impurity could be removed by filtration through a silica plug.
3.1.2 Thioacetate route
The bromo groups of 174 and 176 were converted to thioacetates by reaction with potassium thioacetate using PEG 400 as solvent (scheme 3.1).337 It had been anticipated that selective hydrolysis of the thioester in the presence of the carboxylic ester would be possible. The reaction was attempted using a variety of conditions described in the literature. Under these conditions either no reaction occurred, or cleavage of both the carboxylic ester and thioester was observed. The desired product could not be isolated, although in one instance it was believed to be present in small quantities on the basis of the 1H NMR spectrum. A summary of the reactions and outcomes is given below:
|
177, THF, H2O, Na2CO3, degas, reflux 5 h.189 |
No reaction (by NMR). |
|
175, Acetone, H2O, Na2CO3, degas, reflux 4 h. |
HS(CH2)11OH isolated (by NMR338). |
|
177, Acetone, H2O, Na2CO3, overnight, rt. |
Starting material, traces of HS(CH2)11OH, and desired product. |
|
175, K2CO3, MeCN, Bu4NBr, 19 h.339 |
No reaction (by NMR). |
|
175, NaOMe, MeOH, several min.340 |
Cleavage of ester and thioester (by tlc). |
|
177, 1 eq KOH, EtOH,16 h. |
HS(CH2)11OH and unidentified products. |
|
175, 1 eq NaSMe, MeOH, 30 min, rt.341 |
Mixture. Believed to contain methyl isonicotinate and HS(CH2)11OH (by NMR). |
|
175, 1 eq NaSMe, MeOH, -15 - -10 °C, 30 min, rt.341 |
Mostly starting material. By NMR, equal ratio of CH2OH and CH2SH. |
3.1.3 Thiophosphate route
Thiols may be prepared from alkyl halides by reaction with sodium thiophosphate, to form a phosphorothionate intermediate, which is hydrolysed under neutral or slightly acidic conditions (scheme 3.2).342 The reaction was attempted with 174 in DMF at room temperature, and in refluxing methanol, followed by overnight stirring with water. The starting material was unchanged by the former conditions, whereas the latter resulted in hydrolysis of the carboxylic ester, as judged from the low integration of the CO2CH2 resonance in the 1H NMR spectrum. The hydrolysis may be accelerated by phosphate acting as a base catalyst.
3.1.4 Sodium hydrosulfide method
The classical method of preparation of thiols from alkyl halides involves reaction with a nucleophilic sulfide salt generated by passing H2S through a solution of an alkali metal hydroxide.343 The hydrosulfide salt NaSH·nH2O is now commercially available as a less malodorous alternative to H2S. Reaction of 176 with NaSH·nH2O under aerobic conditions was intended to yield 181, by aerial oxidation of the immediately formed thiol. 1H NMR and ES MS analysis of the reaction products, chromatographically separated into three fractions, indicated successful displacement of bromide but partial cleavage of the carboxylic ester. Each fraction contained molecules with two, one or zero pyridyl groups, but a mixture of sulfur containing functional groups. The sulfur containing products were identified as thioether, disulfide and trisulfide (figure 3.1).344,345 The former arises from nucleophilic attack of thiol on the starting material. Oxidative polymerization of thiols and HS- is responsible for polysulfide products. Due to the extreme similarity of the polarities of the various sulfur products separation on a preparative scale by column or plate chromatography was not feasible. Although it would be possible to avoid formation of these polysulfur products by use of an inert atmosphere, thioether formation and carboxylic ester cleavage are likely to be difficult to avoid.
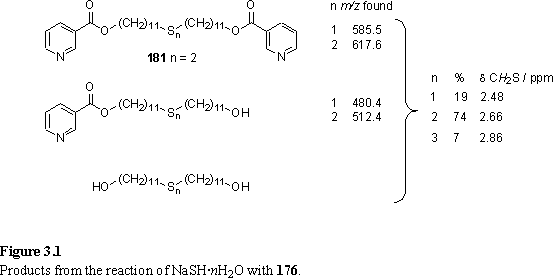
3.1.5 Triphenylsilane thiol method
The original report describing the preparation of triphenylsilane thiol (Ph3SiSH) also described its reaction with alkyl halides to yield Ph3SiSR.346 Recently the use of (Ph3Sn)2S for thiol synthesis from alkyl bromides was described.347 A nucleophilic thiolate is generated by in situ reaction with fluoride anion, which then attacks the alkyl bromide to afford RSSnPh3 which is hydrolysed to a thiol.
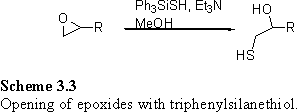
Ph3SiSH in MeOH with triethylamine as a base ring-opens epoxides to yield b-hydroxythiols (scheme 3.3).348 176 treated with Ph3SiSH under these conditions without degassing the solution produced an inseparable mixture of polysulfide species (scheme 3.4) identified by NMR and FAB MS which displayed clusters of peaks separated by m/z 32. Quantitation was possible by integration of the CH2Sn resonances in the 1H NMR spectrum, which revealed the mixture to contain approximately 80 % of the disulfide product and the remainder to be higher polysulfides.
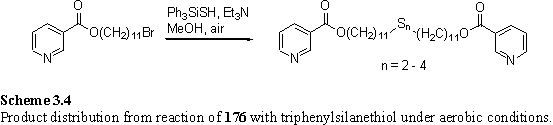
When the reaction was repeated, with initial degassing and use of an inert atmosphere a mixture of starting material, thiol and disulfide was obtained. Of the sulfur containing products greater than 90 mol% was thiol. It is uncertain whether oxidation occurred due to traces of oxygen in the reaction solvent, or during the work-up. It proved impossible to separate the starting material and thiol product by column chromatography so oxidation to the disulfide was carried out by reaction with iodine.349 This resulted in a good separation of the more polar disulfides 180 and 181 from the corresponding starting material. The material thus prepared, in up to 52 % yield, appeared pure by conventional organic chemistry techniques (tlc, NMR, analysis). However when monolayers on Au were prepared using these compounds XPS analysis revealed contaminant peaks attributed to iodine (see section 4.1). It is known that iodide and iodine adsorb to gold,350 and it appeared likely that traces of iodine from the final oxidation step had contaminated the samples of 180 and 181. Therefore a new synthetic procedure was sought which avoided the use of iodine in the final step. Several variants of the reaction with Ph3SiSH were tried, with the aim of causing the reaction to go to completion, or to achieve oxidation without use of iodine. These are summarized below, although none were found to be entirely satisfactory.
|
(i) 174, Ph3SiSH, Et3N, THF, degas; (ii) TBAF, DDQ. |
No product isolated. |
|
(i) 174, Ph3SiSH, Et3N, MeOH, degas; (ii) air. |
Mixture of thiol and 174. Air oxidation too slow to be of use. |
|
(i) 176, Ph3SiSH, Et3N, NaI, THF; (ii) TBAF. |
Impure thiol product. |
|
(i) 176, Ph3SiSH, Et3N, NaI, MeOH, 22 h, rt; (ii) CAN, MeCN. |
181 obtained. Slight impurity by NMR. |
3.1.6 Esterification of disulfanyl bis-(11-undecanol)
Synthetic routes which incorporate a disulfide prior to formation of the carboxylic ester were investigated. This strategy avoids the need to deprotect a thiol whilst avoiding cleavage of the carboxylic ester.
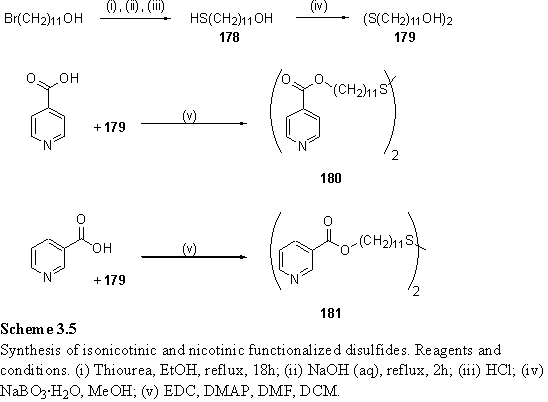
Mercaptoundecanol, 178, was prepared by a modified literature route351 by reaction of bromoundecanol with thiourea, followed by hydrolysis of the thiouronium salt with aqueous NaOH. Oxidation of the thiol to disulfide 179 was performed with sodium perborate352 in aqueous methanol. A variety of conditions were explored for esterification of this diol with nicotinic and isonicotinic acid. The success of the reactions was judged according to the purity of the product as assessed by NMR, elemental analysis and tlc. The conditions and outcomes are summarized below:
|
179, Nicotinoyl chloride·HCl, Et3N, DCM, reflux. |
181, 75 % yield. Slight impurity by elemental analysis. |
|
(i) Isonicotinic acid, SOCl2, reflux; (ii) 179, DCM, Et3N, reflux. |
180, 61 % yield. Good purity |
|
(i) Nicotinic acid, SOCl2, reflux; (ii) 179, Et3N, DCM, reflux. |
181, 78 % yield. Slight impurity by tlc. |
|
Nicotinic acid, 179, DCM, DMF, EDC, DMAP. |
181, 84 % yield. Good purity. |
|
Isonicotinic acid, 179, DCC, DMAP, DMF. |
Incomplete coupling. |
|
Nicotinic acid, 179, DCM, DMF, DCC, DMAP. |
Incomplete coupling. |
|
Isonicotinic acid, 179, EDC, DMF, DCM, DMAP. |
180, 84 % yield. Good purity. |
|
Nicotinic acid, 179, DCM, EDC, DMAP. |
181, 74 % yield. Good purity. |
Reaction of 179 with isonicotinic or nicotinic acid with EDC as coupling reagent and catalytic DMAP in a mixed DCM/DMF solvent afforded the highest yields of pure 180 and 181 (scheme 3.5). It was material produced using this procedure that was used for the surface chemistry experiments described in the following chapter.
The crystal structure of 181 was obtained (figure 3.2) and the molecular packing is of interest in relation to the potential packing of the molecule as a SAM on Au(111). The molecules crystallized in the unusual space group P1, so the crystals lack a centre of symmetry even though the molecular structure lacks any intrinsic chirality. As expected from ab initio calculations of alkyl disulfides described in the literature,353 the disulfide bond is in a gauche conformation with a C-S-S-C torsion angle of 68.8(5)°.

The alkyl chains are in an all trans conformation, and are approximately close packed (figure 3.3).

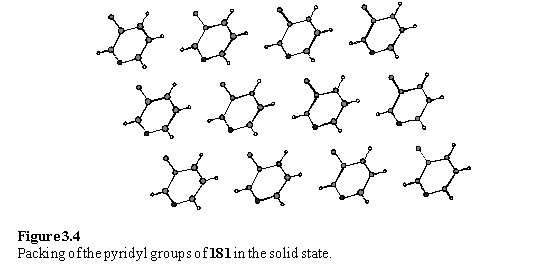
If a plane is placed through the sulfur atoms a two dimensional lattice is obtained with primitive lattice parameters of 5.4 Å, 4.9 Å and angle of 71°. A well packed monolayer of simple alkane thiols on Au(111) has a hexagonal lattice with a lattice parameter of 5.0 Å, which suggests that 181 is not shaped to pack ideally on Au(111). However the literature does describe thiols and disulfides with elliptical cross sections forming monolayers on Au(111) with non-hexagonal lattices.18 If a slice through the crystal structure of 181 can be considered a model for a monolayer then the nitrogen atoms of the pyridyl groups lie in the plane of the layer (figure 3.4) suggesting that they would be inaccessible for coordination to metals without reorganization of the layer.
3.1.7 Quinuclidine ligands
A thiol appended amine donor with a different metalloporphyrin binding constant to pyridine was desired for comparison with 180 and 181. Quinuclidine was chosen as the nitrogen ligand, because the amine was expected to bind more tightly than pyridine to metalloporphyrins.282 The same design criteria were used as for pyridyl ligands, namely attachment of a C11 alkyl chain terminated in a thiol or disulfide by means of an ester or ether linkage. Several synthetic routes starting from commercially available quinuclidine derivatives were explored and these are described in the following sections.
3.1.8 From 4-cyanoquinuclidine
Direct alcoholysis of 4-cyanoquinuclidine with bromoundecanol catalysed by sulfuric acid was not successful.354 Precipitation of the sulfate salt of 4-cyanoquinuclidine occurred and this proved difficult to solubilize in organic solvents. Conversion of the 4-cyanoquinuclidine to an ethyl ester, suitable for elaboration by transesterification met with partial success using HCl in EtOH over 6 days at room temperature.355 The 1H NMR spectrum of the HCl salt of the products in D2O showed resonances at 1.27 and 4.21 ppm attributed to an ethyl ester. Unfortunately the product was contaminated with an unidentified impurity, visible by tlc and NMR, which proved difficult to separate.
Attempts were made to react the 4-cyanoquinuclidine with a Grignard reagent prepared from undecenyl bromide (scheme 3.6). The reaction was carried out in refluxing ether, or toluene with 1 eq ether356 but in neither case was the desired ketone isolated, although under the former conditions LC MS analysis indicated the presence of the product by a peak at m/z 292.
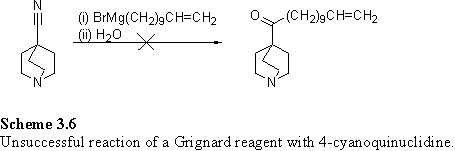
3.1.9 Alkylation of 3-quinuclidinol
An alternative scheme involving alkylation of 3-quinuclidinol with a long chain terminal alkene, followed by free radical addition of Ph3SiSH357 or AcSH358 to the alkene is shown in scheme 3.7. Attempts at selectively alkylating the oxygen of 3-quinuclidinol without protection of nitrogen were not successful. Treatment of 3-quinuclidinol with NaH in THF followed by reaction with undecenyl bromide afforded a major product which, by the integrals of the 1H NMR spectrum, appeared to contain 2 alkene units per molecule. The other resonances did not resemble those of 3-quinuclidinol indicating that the quinuclidine moiety had fragmented. Electrospray mass spectrometry also supported the hypothesis of double alkylation, with the major product giving a molecular ion peak at m/z 432. The literature describes the use of borane as a protecting group for the nitrogen of quinuclidine during etherification,359 although this was not pursued due to success of esterification reactions discussed in the next section.
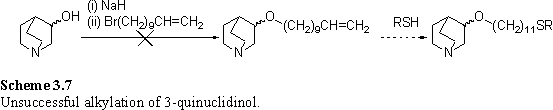
3.1.10 Esterification of 3-quinuclidinol
From the experiences of alkylation of 3-quinuclidinol and the observation of ready alkylation at nitrogen, it was decided to avoid coupling 3-quinuclidinol to alkyl carboxylic acids with a terminal alkyl halide group, as this was predicted to lead to polymer formation.
Two routes were considered, esterification with undecenoic acid followed by conversion of the alkene to thiol, or incorporation of the thiol, protected as a thioacetate prior to the esterification. The latter route was chosen as this requires fewer steps to be carried out on the amine containing species. The quinuclidine derivatives were found to be difficult to purify due to extensive streaking during column chromatography.
Acid 182 was easily prepared by reaction of bromododecanoic acid with potassium thioacetate, and could conveniently be purified by recrystallization. The acid was coupled with racemic 3-quinuclidinol using EDC with DMAP as nucleophilic catalyst (scheme 3.8). Avoidance of chlorinated solvents during the work-up was found to be essential to avoid emulsion formation. Evaporation of the reaction solvent and redissolution in EA, followed by a standard aqueous work-up prevented this problem and 183 could be isolated in a mediocre but acceptable yield of 53 %.
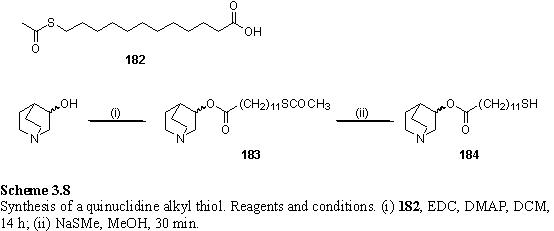
For selective removal of the thioacetate protecting group, without cleaving the carboxylic ester, the reaction with sodium thiomethoxide in MeOH was chosen.341 This has the advantage that the thiolate also acts as a reducing agent, inhibiting the formation of disulfides by atmospheric oxidation.
Reaction of 183 with 1 eq NaSMe in MeOH with the temperature increasing from -15 - -10 °C over 30 minutes produced a chromatographically inseparable mixture of starting material and 184, as judged by the 1H NMR spectrum. Disulfide oxidation products were not observed. It was found essential to ensure that the reaction had proceeded to completion by use of 1.2 eq NaSMe in degassed MeOH at room temperature for 30 minutes. Analytically pure 184 was obtained after chromatography and hot filtration in hexane solution to remove residues probably arising from elution of the silica column with a polar solvent mixture (CHCl3/MeOH). Enantiomerically pure 184 was prepared by the same route, but starting from homochiral (R) (-) 3-quinuclidinol. Although the enantiomerically pure compound will have the same binding constant to achiral metalloporphyrins as the racemic mixture (in the absence of self association of 184 in solution), packing of racemic and homochiral compounds in a SAM is not necessarily the same. The racemate may undergo a spontaneous two dimensional resolution on the surface360 into domains of single enantiomers, or form a homogenous structure consisting of a mixture of both enantiomers.
3.1.11 Model compounds
Model compounds lacking the mildly air sensitive and potentially metal coordinating thiol or disulfide group were required to investigate the binding of these ligand to metalloporphyrins in solution. Nicotinoyl and isonicotinoyl esters 140 and 185 were prepared from acid chlorides, and purified by precipitation of the sulfate salt from ether. Similarly racemic 186 was synthesized by reaction of 3-quinuclidinol with decanoyl chloride.