2.1 Preparation of Rh(III) diaryl porphyrin
5,15-Diaryl porphyrin was synthesized according to a published procedure (scheme 2.2).228 Pyrrole derivative 115 was prepared up to a 50 g scale by a lead tetraacetate oxidation, and acid catalysed self condensation yielded protected dipyrromethane 116. Debenzylation by catalytic hydrogenolysis with Pd/C followed by decarboxylation with TFA afforded a dipyrromethane intermediate which was not isolated but immediately condensed with benzaldehyde. The resulting porphyrinogen was oxidized with DDQ to yield crude 117 which could be purified by a combination of filtration through a silica column and recrystallization. Batches of greater than 1 g of pure 117 in up to 70 % yield could conveniently be obtained using this procedure.
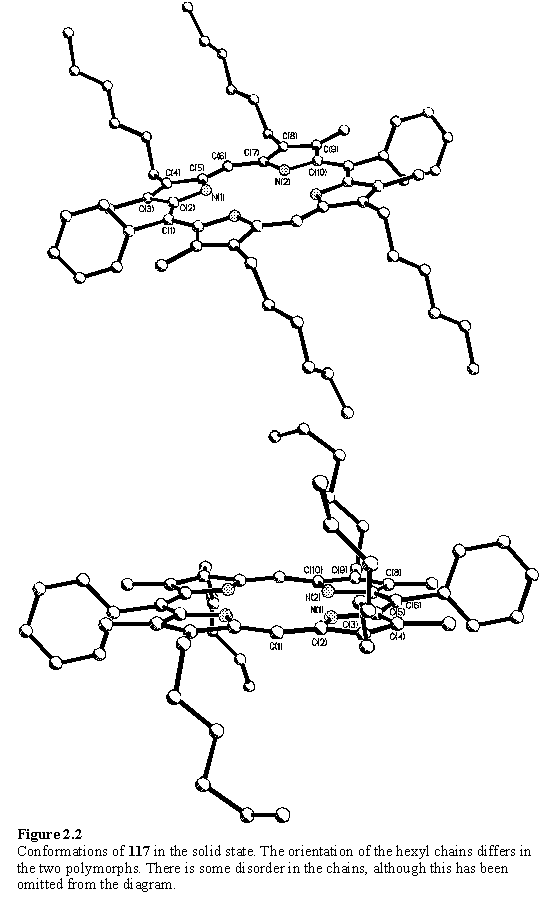
Attempts at growing X-ray quality crystals of 117 afforded crystals with differing external morphology. Both fine purple needles and red plates could be obtained from the same crystallization batch, with the needle form predominant. There is ample precedent229 for different polymorphs of a compound to crystallize under apparently identical conditions. X-ray analysis was performed on two types of crystal, although these were not grown under identical solvent conditions. The analysis revealed that these were both solvent free and hence true polymorphs, the structures (figure 2.2) differing in the orientation of the hexyl chains with respect to the plane of the porphyrin, and as a consequence the molecular packing. The porphyrin core is elongated along the aryl-aryl axis, a feature which is common to free-base porphyrins with this substitution pattern.230 The distortion is of the same magnitude in each of the polymorphs, the N - N distances along the Ar-Ar axis being 3.181(7) and 3.195(3) Å, whereas along the unsubstituted meso axis the N - N distances are 2.719(9) and 2.702(3) Å. The flexible hexyl groups are disordered, a feature which is common to porphyrins of this type.103 However the use of unsubstituted phenyl groups at the 5 and 15 positions of the porphyrin instead of bulky 3,5-di-tert-butyl-phenyl as in reference 103 avoids disorder associated with the tertiary butyl groups, introduced to increase solubility. The solubilizing effect of the hexyl substituents of 117 was found to be acceptable and all derivatives prepared from 117 displayed good solubility in chlorinated solvents.
Rhodium metallation of 117 was carried out using a modified literature procedure231 by treatment with Rh2(CO)4Cl2 and sodium acetate in degassed DCM, followed by iodine oxidation. During the first step of the reaction the red solution became deep green, and this has been reported to be due to formation of intermediates with a pair of Rh(CO)2 fragments on opposite faces of the porphyrin coordinated to adjacent pyrrole nitrogen atoms.232-234 After purification by column chromatography pure 118 was obtained as a brown solid which readily dissolved in chlorinated solvents to form deep orange solutions. Yields for the metallation reaction were in the range 44 - 89 %, and appeared to depend on the quality of the Rh2(CO)4Cl2 starting material, which degrades on storage. The use of dry and degassed solvent was also believed to be important in ensuring the success of the reaction.
Recrystallization of 118 from solutions containing methanol yielded almost black crystals, which dissolved only slowly in chloroform, although sonication greatly increased the dissolution rate. 1H NMR spectra of samples of this material in CDCl3 exhibited a resonance at 10.30 ppm, attributed to the meso protons, and a pronounced splitting of the hexyl methylene H1 resonance into a pair of multiplets at 4.02 and 3.90 ppm. The latter splitting is due to the two faces of the porphyrin being rendered inequivalent by coordination of the axial iodo ligand. Typically a 1H broadened quartet was observed in the range -2 - -2.4 ppm and this was assigned to the hydroxyl proton of an axially coordinated methanol molecule, introduced during the recrystallization step, which completes the coordination sphere of the rhodium. This solvent remains coordinated in the solid state, even after extensive drying of the crystals in vacuo and months of storage. The methanol CH3 resonance was usually obscured by the more intense hexyl methylene resonances at 1.36 - 2.22 ppm. Addition of oven dried 4Å molecule sieves to the NMR sample caused the OH resonance to shift to -2.48 ppm, and the CH3 resonance appeared at 1.02 ppm. After standing overnight both peaks vanished as the methanol was absorbed by the sieves. The chemical shift of the meso resonance remained unchanged. In a further experiment titration of a solution of 118·MeOH in CDCl3 with methanol resulted in downfield shifts of both the OH and CH3 resonances, consistent with an exchange of free and bound methanol which is fast on the NMR chemical shift time-scale. A peak, initially at 1.17 ppm, was also observed to be downfield shifted during this titration and this was assigned to water which competes with methanol for coordination to the rhodium centre. This precluded a quantitative analysis of the titration data, as the amount of water present and its binding constant to the porphyrin are unknown. These results are also consistent with the upfield shifts of the MeOH resonances observed upon addition of molecular sieves, in which case the chemical shifts tend towards those of the 118·MeOH complex.
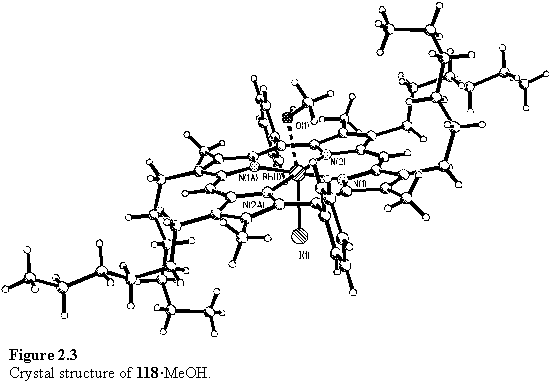
Crystals of 118 suitable for X-ray diffraction were obtained from a DCM/methanol solvent mixture. The structure is shown in figure 2.3 and confirmed the hypothesis, made on the basis of the NMR evidence, that a methanol molecule coordinates to the rhodium centre. The methanol and iodo ligands are disordered over an inversion centre on the Rh atom, with a Rh-O bond length of 2.201(15) Å. The porphyrin itself shows little deviation from planarity, with an rms deviation of 0.03 Å from the best fit plane.
Recrystallized 118 with coordinated methanol was used as the starting point for many of the experiments described in the following sections. It has the advantage of a well defined composition, as opposed to material possessing an unknown ‘solvent’ ligand at the sixth coordination site of the metal.
Under some circumstances the presence of methanol could be detrimental, due to the mild acidity of the hydroxyl proton, so as an alternative the preparation of a complex with the more inert THF ligand was investigated. After chromatography of 118 the resulting solids were dissolved in THF and the solvent evaporated followed by drying in vacuo. The NMR spectrum of the material so obtained, displayed an approximately 8H broadened peak with a shoulder at 0.42 ppm which can be assigned to the overlapping resonances of weakly coordinated THF. In practice, the 118·THF complex was less preferred to 118·MeOH for NMR experiments involving displacement of these labile ligands, as the free THF resonances at 3.76 and 1.85 ppm tended to partially obscure peaks in these regions of the spectrum, whereas free methanol did not cause such interference.
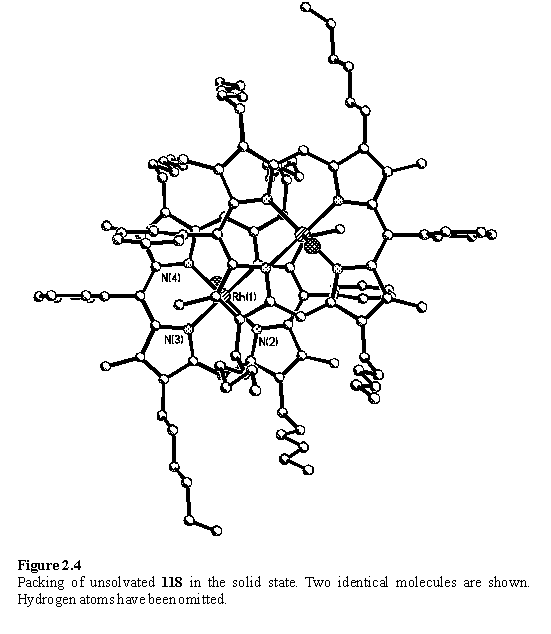
In an attempt to organize a dimeric porphyrin architecture in the solid state utilizing Rh coordination to diols, crystallizations were set up in which DCM solutions of 118 were layered with ethylene glycol and propylene glycol. These diols are immiscible with DCM but after several months, during which time the volume of DCM progressively reduced due to evaporative losses, crystals were recovered from the DCM/ethylene glycol sample. Surprisingly, the structure (figure 2.4) consisted of tightly p stacked dimers of porphyrin in which the rhodium is only five coordinate. The absence of the sixth ligand permits the porphyrins to approach to a separation of only 3.2 Å in the offset geometry predicted for p-p stacking.235 Evidently there is a balance between weak coordination of alcohol and p stacking in the solid state, an effect which has been reported for zinc porphyrins crystallized from methanol containing solvents.236 This is not a preparatively useful route to solvent free 118, due to the length of time taken to obtain crystals, and microanalysis of the remaining solids not used for crystallographic analysis was not consistent with a homogenous composition.